Shortly after the 2017 release of the European Atherosclerosis Society’s (EAS) consensus statement deeming low-density lipoprotein cholesterol (LDL-c) causal in atherosclerotic cardiovascular disease (ASCVD), a group of authors (Ravnskov et al.) published a “comprehensive review” that attempted to falsify the EAS paper’s conclusions1,2. This paper is frequently cited and appears to lend considerable weight to arguments put forth by self-proclaimed lipid-hypothesis skeptics, but this couldn’t be further from the truth. Given the immense negative implications that inaccurate advice pertinent to cardiovascular disease (CVD) possesses, this article intends to comment on the incoherent, misleading, and patently false nature of the declarations in the Ravnskov et al. review. Hopefully the information provided in this article will discourage the adoption and propagation of harmful views posited by these authors going forward.
Section 4.1
Although Ravnskov et al. begin with a few sections on total cholesterol and CVD, this will not be discussed directly because the main focus of the original EAS consensus statement was the role of LDL-c in CVD. Thus, Ravnskov et al.’s first LDL-c specific contentions are that the current evidence supporting LDL-c causing CVD is based on “select patient groups”, and there is evidence suggesting higher LDL-c does not always translate to more atherosclerosis. But as I will clarify, the former contention is false, and the latter view is reductionist. Not only does Ravnskov et al. make generalizations about causation that completely ignores the multifactorial nature of CVD here, but we will also see that the only rash conclusions made from select patient groups are from Ravnskov et al.
To support their position about select patient groups, Ravnskov et al. cite four studies with (supposedly) a lack of an association between LDL-c and the degree of atherosclerosis. The first study is a bare-bones case report of 123 people who died in car crashes3. The results are based on undisclosed biomarker measures of randomly selected death cases that don’t represent any particular group. There are virtually no details given about serum cholesterol or the prevalence of atherosclerosis in these subjects, so drawing conclusions from this sample is extraordinarily naive.
The following citation leads to a similar problem, including a selection of around 200 case-studies of individuals who died from accidental causes, with no details on other risk factors provided4. It simply concluded that there was no correlation between total cholesterol and the extent of atherosclerosis. Yet, only sentences later, Mathur et al. state, “When all the cases were divided into arbitrary groups according to the amount of atherosclerosis, a rise in the levels of mean serum total cholesterol was seen in the first six successive groups of aortic atherosclerosis.”. This change in atherosclerosis progression from serum cholesterol suggests that there was a relationship. The authors mention that the effect was attenuated when adjusted for age yet ignore the dozens of other equally plausible confounders. The lack of consideration for other confounders is enough in and of itself to deem this study one of poor quality when analysing causal relationships.
The third study is yet another collection of case reports from 58 individuals aged 60-69 years old. Much like the previous studies discussed, measures of serum cholesterol were again compared with the extent of atherosclerosis in various arteries5. The researchers did not adjust for any confounders nor consider the temporality and cumulative exposure of total cholesterol; or more importantly, LDL-c. So, unsurprisingly, they did not find a correlation between total cholesterol and atherosclerotic burden. The study authors did, however, note that before excluding individuals with values of total cholesterol over 300 mg/dL, a trend was indeed present. That being said, this trend would still be remarkably tenuous evidence to support any strong position.
The fourth and final study that Ravnskov et al. cite is, shockingly, another case study of 43 individuals who died suddenly from various causes6. It contains the same limitations as the previous three and once again focused on total cholesterol instead of LDL-c, “failing” to find an association with the severity of atherosclerosis.
After these four case studies, Ravnskov et al. refer to a study of 304 asymptomatic women that supposedly demonstrates no association between LDL-c and coronary artery calcification, as measured via electron beam tomography (EBT)7. Notably, this paper intended to observe coronary artery disease risk prediction capacity for the National Cholesterol Education Program (NCEP) ATP-II lipid guidelines, not the relationship between LDL-c and calcified plaque. Despite this, the study still clearly showed those under 55 years old with positive EBT scans (indicating calcified plaque) had significantly higher LDL-c. Granted, there was a lack of association for those over 55 years old. However, given that calcified plaques are highly correlated with age, we should not expect major differences to be observed unless there are considerable contrasts in LDL-c among the older patients. There were not. Reflected in their third table, the mean LDL-c values for positive and negative women were very similar. The women with positive EBT scans did have higher LDL-c, just not significantly greater. To add, we should again note that the lipid values are acute measurements and do not appropriately consider the overarching issue: cumulative LDL-c exposure.
In quite a hilarious turn of fate, Ravnskov et al. then mention results from “one exception” study, which is on a subcohort of individuals in the Progression of Early Subclinical Atherosclerosis cohort. I say hilarious because this study is infinitely higher quality than the other compilations of poorly detailed autopsy case-studies they cited previously8. Their results revealed that as LDL-c (measured prospectively at three time periods) increases among those with all other major known risk markers in the normal range, so does the presence and extent of atherosclerosis, reflecting the existence of a temporal, dose-response relationship that Ravnskov et al.declares doesn’t.
Ravnskov et al. response to these findings are, “However, association does not prove causation. Mental stress, for instance, can raise cholesterol by 10–50% in the course of half an hour, and mental stress may cause atherosclerosis by mechanisms other than an increase in LDL-c; for instance, via hypertension and increased platelet aggregation.” This statement is rather strange because, while Ravnskov et al. are clearly comfortable supporting earlier points using poorly detailed collections of incredibly old case studies that don’t consider LDL-c and the multifactorial nature of CVD, they now suddenly dismiss results from a larger and more comprehensive prospective cohort that considers temporality, exposure-response, and other known risk factors because “correlation doesn’t equal causation”. The line of argumentation is inconsistent. Also worth noting, this is far from the only study of its kind. There are many more similar studies to the Progression of Early Subclinical Atherosclerosis cohort, yet they don’t disclose them and instead act as if this is an outlier.
Furthermore, Ravnskov et al. mention that mental stress can acutely raise cholesterol 10-50% within a half-hour, then somehow conclude mental stress may cause CVD via a mechanism other than changes in cholesterol, such as hypertension and increased platelet aggregation. Again, given the LDL-cVD relationship is temporal, cumulative exposure is what matters, not minor acute fluctuations. Ravnskov et al. do not even provide evidence for the “mechanisms other than an increase in LDL-c” that supposedly cause CVD, despite hypertension being directly addressed in the PESA cohort they had just cited. It also seems rather strange that Ravnskov et al. are now ironically rejecting claims about a lack of correlation between serum cholesterol and the degree of atherosclerosis because of a lack of consideration for other factors, when they just cited multiple autopsy studies that do not consider additional risk mediators. Inconsistent lines of argumentation, once again.
Section 5.1
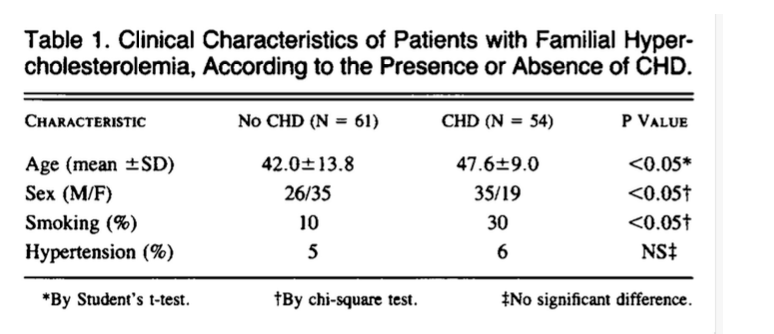
Following the false claim that the relationship between LDL-c and CVD was founded upon select patient groups, Ravnskov et al. reason that if LDL-c causes CVD, serum LDL-c in untreated patients with the condition should be higher than normal. This point isn’t sound, though, as it ignores the patients’ lipid history in the decades before diagnosis, the presence of additional risk factors that can further modulate CVD risk, and the possibility that patients may have adopted lifestyle changes to reduce their risk following a health scare and/or abnormal test results. Regardless, the research Ravnskov et al. cite still fails to solidify their argument. They mention an American study of over 140,000 patients with acute myocardial infarction with “lower than normal” LDL-c at admission. They also mention another study with a similar finding where mortality was “twice as high” in those with LDL-c under 105 mg/dL compared to those with higher values after three years of attempting to lower its concentration9,10.
Regarding the first reference, the mean LDL-c value was 104 mg/dL. This value might be “lower” than the population reference values, but it is far from low. We know from the PESA cohort that this LDL-c value allows for the progression of ASCVD even in the complete absence of other risk factors like smoking, hypertension, impaired kidney function, diabetes, and so on; which were still present in 30.4, 54.2, 6.5, and 26 percent of this sample population, respectively. Additionally, over 21% of the sample used lipid-lowering medication, which inevitably pulls the mean LDL-c downward.
Lastly, it is well-acknowledged by researchers that LDL-c decreases after 24 and up to 48 hours following an acute myocardial infarction (MI) and that the magnitude of the changes is highly correlated with the presence of diabetes11,12. Such a phenomenon emphasizes that post-MI LDL-c values are likely unreliable when analysing the cumulative effects of LDL-c exposure on CVD risk.
The point regarding diabetes exacerbating the reduction in LDL-c observed is crucial concerning the second study Ravnskov et al. referenced, in which lower LDL-c (<105 mg/dL) following non-ST segment elevation MI (NSTEMI) was associated with much higher mortality after three years. In addition to the diabetes consideration, the authors of this paper also note that previous research has demonstrated the severity of the CVD event influences the magnitude of the decrease. Reviewing the baseline characteristics of the patients in this study, just about every non-LDL-c risk factor was less favorable in the lower LDL-c group than in those with higher concentrations. Among them, the percentage with a history of MI, diabetes, hypertension, peripheral vascular disease, and on multiple medications were significantly greater, and HDL was significantly lower.
Even in the conclusion of the actual study, the authors caution against drawing strong conclusions from the data as, despite adjusting for most of the key group differences, residual confounding was still possible if other confounders were not appropriately accounted for. The authors also emphasize the uncertainty of the effect of MI on the lipid profiles in the first 24 hours post-admission, and that the selective focus on NSTEMI may further limit the external validity of their findings. It appears as though Ravnskov et al. may have ignored these concerns from the study authors.
Another issue to add that was not mentioned by the study authors is the potential for reverse causality. We know that conditions such as cancer and infections can reduce LDL-c, and therefore the lower LDL-c values may be a product, not a cause, of potentially fatal health outcomes. Ravnskov et al. reason that reverse causality is not true because it’s more likely that infections cause CVD. Their supporting evidence for this claim is that LDL-c inactivates “almost all types of microorganisms and their toxic products”. They also claim that individuals with low LDL-c have a significantly increased risk of infectious disease and cancer. Sure, they cite three studies to justify these claims, all of which are authored by Ravnskov. Two of them are editorials peppered with mechanistic speculations, and the other is a review of observational studies and statin RCTs that supposedly shows lower cholesterol and/or statin use increases cancer incidence13. However, most, if not all of the RCTs discussed in the review, had incredibly low incidences of cancer/infections. Even the RCTs which noted significant differences between groups hardly break single-digit case number differences despite a low total number of cases. Some of the other RCTs did not demonstrate a significantly increased risk, and the massive confidence intervals indicate low certainty in the LDL-cancer relationship.
The 9 human cohort studies in the review looked at a completely arbitrary selection of cancers (many of which weren’t even identified). Only two suggested the risk of cancer mortality was increased significantly from low LDL-c in both men and women. None of the cohorts suggested cancer incidence was increased in both sexes. The adjustment models used in these cohort studies were also embarrassingly insufficient to say the least. Of the six cohorts for which adjustments were characterized, three did not adjust for alcohol intake, 1 did not adjust for smoking, and another did not adjust for age. This is completely unacceptable for analyses looking at the relationship between an exposure and cancer risk when considering that all these external factors are all risk mediators for the disease.
Contrary to the small collection of observational studies and RCTs discussed in Ravnskov’s editorial, much more comprehensive meta-analyses of RCTs and longer observational studies have shown statin treatment does not increase the risk of cancer or infection incidence/mortality14-17. These were all left out of Ravnskov et al. paper.
On treatment LDL-C and Cancer Incidence
(Alsheikh-Ali et al. 2008)
Effect of Statins on Cancer Incidence
(Cholesterol Treatment Trialists’ (CTT) Collaboration et al. 2012) Effect of Statins on Cancer Mortality
(Cholesterol Treatment Trialists’ (CTT) Collaboration et al. 2012)
Effect of Statins on Cancer Incidence by Site
(Cholesterol Treatment Trialists’ (CTT) Collaboration et al. 2012)
Cancer Mortality, Recurrence, Progression + Disease-Free Survival (Jeong et al. 2020)
(Van den Hoek et al. 2011)
(Van den Hoek et al. 2011)
Shifting the discussion to infections, a large cohort study including a sample of individuals who were admitted to the hospital for infection revealed that when comorbidities are taken into account, lower genetically predicted LDL-c concentrations did not increase SEPSIS risk18. Subsequently, the authors concluded lower LDL-c did not appear to inherently increase the risk of the condition and that comorbidities were likely responsible for any increase in risk attributed to lower LDL-c in other studies.
(Feng et al. 2019)
Similar results were obtained in another large study investigating the influence of lipoproteins on the risk of overall infectious disease19.
(Trinder et al. 2019)
Finally, Mendelian randomization (MR), a tool far better suited to determine causal relationships, demonstrates that acute and lifelong genetically determined lower LDL-c does not increase the risk of cancer20. As seen below, an observational analysis from Benn et al. showed that an association was present between LDL-c and cancer risk in a subset of their sample, but not in those who had lower LDL-c due to genetic polymorphisms, clearly indicating reverse confounding is at play despite Ravnskov et al.’s claims otherwise.
In closing, the arguments offered by Ravnskov et al. in this section are based solely on a selection of small, poor-quality, and likely confounded studies. These studies are vehemently opposed by the results of multiple larger, higher-quality investigations which Ravnskov et al. ignored or chose to dismiss.
Section 5.2
In this brief section, Ravnskov et al. remark that if LDL-c were the primary cause of ASCVD, then people with the highest LDL-c values would have shorter lives than people with lower values. While at face value this appears to be a reasonable claim, it is haphazard in reality. Though ASCVD is a major cause of death worldwide, there are an infinite number of other potential causes of death, and CVD itself takes decades to develop before causing an event or death. Ravnskov et al.’s assumption that high LDL-c values (especially acute measures at a single time point) would always lead to shorter lives disregards the multifactorial nature of CVD entirely. Not only that, but the meta-analysis (Ravnskov’s publication) and cohort (which comprised 2/3rd of the entire meta-analysis’ sample) they posit to demonstrate this is remarkably biased. The meta-analysis is saturated with major methodological pitfalls and further demonstrates ignorance of numerous other factors that are critical to consider when characterizing the relationship between LDL-c and CVD or all-cause mortality21,22. Expectedly, the publications that Ravnskov et al. cite have received several detailed, evidence-based replies identifying many of these issues. These replies clarify why that the conclusions drawn from the meta-analyses are entirely inappropriate. Two of the best responses were from David L. Keller and Eatz et al., covering the false assumption that the association between TC or LDL-c and CVD mortality should be the same in the elderly as younger individuals. Such false assumptions are usually due to:
Failure to consider other known mortality risk factors
Lack of consideration for survivorship bias and reverse causation
Insufficient detail on the multivariate adjustment models
Not accounting for statin use
Ignorance of RCTs demonstrating benefits of statins on mortality in the elderly.
Eatz et al. add that a Mendelian randomization (MR) study carried out by Postmus et al. revealed a genetic predisposition to higher LDL-c contributes to a substantial increase in mortality, even in the oldest samples (>90 years old)23.
A more recent MR provides further support for this point, revealing that genetically predicted higher apolipoprotein B (apoB – a protein present on LDL-c particles and whose concentration predominantly depends on that of theirs) and LDL-c were associated with decreased lifespan and a much lower chance of reaching the 90th percentile of lifespan. Multivariable estimates indicate the relationship is explicitly driven by the former (apoB)24.
Overall, there is a profound lack of actual support for the main claim made in this section of the Ravnskov et al. paper. The “evidence” brought forth to assert that higher LDL-c does not correspond with increased mortality is founded on a collection of poor-quality, confounded data sets. They fail to account for many factors that invalidate their argument while exerting willful ignorance of a large body of higher quality evidence refuting their position.
Section 5.3
In what has to be the lowest effort to dismiss strong evidence for the causal role of LDL-c in CVD within the entire article, Ravnskov et al. say that while lower genetically predicted LDL-c in MR studies is associated with lower all-cause mortality, association does not equal causation. This is an odd point given the article is meant to focus on CVD, but I will comment anyway. Ravnskov et al. continues to state, “Other genes in the same individual may have opposite effects, and as pointed out by Burgess et al., ‘Power, linkage disequilibrium, pleiotropy, canalization and population stratification have all been recognized as potential flaws in the Mendelian randomization approach”. This claim is an incredibly dishonest and remarkably weak generalization. The reference they provide for this is a 2015 review of the current state of MR, including a discussion of potential issues and recent advances to overcome these issues25. What is strange is that the cited paper concludes that while some MR issues may have gone unrecognized in the past, they have become far more acknowledged. Many new methodological improvements have arisen to combat influencers that may have distorted past results. The cited paper goes on to say that these methodological improvements allow for more reliable conclusions about causal relationships to be drawn using MR research.
This message contrasts with the extreme skepticism and dismissal that Ravnskov et al. puts forth, demonstrating a grossly selective interpretation of the actual content. It is hard to imagine this misinterpretation was anything but intentional in order to fit their narrative. Accompanying the existing collection of MR research, more analyses have surfaced in recent years that are highly concordant with those of the past, including an excellent study from last year that included a massive selection of single nucleotide polymorphisms (SNPs) that alter LDL-c.
So, all in all, contrary to the claims of Ravnskov et al., the amount and consistency of MR findings, including incredibly robust analyses of numerous SNPs affecting LDL-c, show that LDL-c changes correspond directly to changes in CVD risk26. Attempts to dismiss these results can best be likened to someone furiously scooping buckets full of water out of the ocean and subsequently claiming to have drained it.
Section 6.1
Ravnskov et al. starts this section off with a simple point that is partially valid but then follows up with a convoluted collection of incomprehensible remarks, cherry-picked evidence, and conveniently sweeps crucial details under the rug. They begin by saying the most decisive proof of causality is when the lowering or elimination of a suspected causal factor results in a lower incidence of the disease in question. They follow this by admitting there have been “small statistically significant benefits” in coronary event outcomes from statin trials, but they are quick to question if these statin benefits are actually from lowering LDL-c. While the first point about the best causality evidence for causality is mostly agreeable, it still fails to consider important nuances such as cumulative LDL-c exposure and other risk mediators. The second point is extremely odd, because the trials that Ravnskov et al. proceed to discuss reveal substantial CVD risk reductions with greater LDL-c reduction, with more marked risk reductions when statin therapy is initiated earlier and maintained for a longer duration. Ravnskov et al. continue to say that three reviews (including the one this paper is supposedly a rebuttal to) claim that statin trials demonstrate an exposure-response when comparing the CVD outcomes to the degree of LDL-c lowering achieved, but that “it is impossible to know whether the greater effect of a trial using a higher statin dose may be caused by its cholesterol-lowering effect or pleiotropic effects”. Further, they state, “True exposure–response is based on a comparison between the degree of cholesterol-lowering in each patient in a single trial and the absolute reduction of their risk. True exposure–response has only been calculated in three clinical statin trials, and it was absent in all three.”
Their position regarding uncertainty if a higher degree of LDL-c lowering was the cause of the reductions in CVD incidence observed in statin trials is one of unfettered skepticism with no solid basis. Especially in the case that the variable being investigated as causal tracks very strongly (in an exposure-response manner) with changes in CVD risk across dozens of investigations, the chances that any other ostensibly unknown variable happens to possess the same (or better) correlation is astoundingly low. Even just the aforementioned MR studies illustrate remarkably consistent results concerning the relationship between LDL-c (or even better, apoB) and CVD risk across dozens of single nucleotide polymorphisms in a dose-response manner. This alone indicates the chances of pleiotropic effects being responsible for the LDL-cVD relationship are infitessimately small. The statin reviews that I discussed at the beginning of this section further reinforce this point as they demonstrate the achieved risk reduction for CVD correlates near perfectly with the the difference in LDL-c achieved between the control and intervention group, regardless of the method utilized, in RCTs, MR, and observational research.
As for the second point, it’s not entirely clear what Ravnskov et al. are trying to suggest. It seems as if they’re defining exposure-response as the mean change in absolute risk of CVD seen in each patient according to the degree of cholesterol-lowering they experienced, rather than relative change in CVD risk. They declare that this “true exposure-response” has only been calculated in three trials. Yet, only one of these three trials explicitly stated an absolute risk reduction value. Regardless, whether they are discussing relative or absolute CVD risk changes, the fact remains that they are using only three old and far less methodologically rigorous trials in an attempt to disprove the findings from multiple MR studies and RCT meta-analyses of statin and non-statin LDL-c-lowering therapies, which demonstrate a clear dose-response effect on CVD risk. This is nothing less than gross negligence of appropriate scientific methods and the hierarchy of evidence.
All that said, even if one were to ignore this abuse of proper scientific methodology completely, their sources don’t provide strong support for their claims. The first study that Ravnskov et al. claim showed a lack of “true exposure-response” is a secondary analysis of the West of Scotland Coronary Prevention Study Group trial27. This trial was initially conducted to investigate the influence of treatment with pravastatin for five years on clinical CVD events in moderately hypercholesterolemic men aged 45 to 64. Immediately worth noting is that given the nature of this analysis. The study authors explicitly stated, “The results described here are derived from post hoc analysis and therefore must be viewed cautiously”, something Ravnskov conveniently left out. That being said, it is also apparent when delving into the results of the study that there are many potential sources of error that render Ravnskov’s takeaway as uncharitable at best. These sources of error are not the authors’ fault as there were many limitations in the knowledge that would have existed when the trial was carried out, but they make for an essential point in this discussion. Specifically, the study found that LDL-c reduction elicited a significant decrease in the rate of CHD, but that the maximal benefit of statin therapy (an ~45% relative risk reduction in CHD) was achieved with a 24% reduction in LDL-c, and that absolute LDL-c reduction did not correlate linearly with the decrease in CHD risk.
However, there are quite a few things worth discussing that could have diminished the accuracy of the finding specific to absolute LDL-c reduction. Most notably, the risk reduction attributed to absolute LDL-c lowering was representative of the CHD risk differences observed between the control group (with a certain mean LDL-c) and the intervention group that experienced the specified LDL-c reduction. While it may be expected that measures of absolute LDL-c reductions would represent the difference in achieved LDL-c between the two groups–thus giving a picture of how CHD risk tracks with LDL-c change–this might not have been the case. If there was a considerable variance in baseline LDL-c concentrations of subjects in the intervention group, it’s possible those with higher baseline concentrations, despite experiencing an appreciable absolute LDL-c reduction, may still end up with a final concentration similar to the final mean LDL-c of the comparator control group. Even just the existence of a small number of subjects in the intervention group for which this occurred could impinge the detection of a dose-response relative risk reduction. If their final LDL-c concentration weren’t much different from the mean LDL-c value in the control group, their risk wouldn’t be either. This may seem absurd to suggest given that randomization should prevent this sort of skewed distribution, but it is worth considering since the sample for this particular analysis was not actually randomized and included only a subset of subjects who achieved over a 5% reduction in LDL-c (2600 of the 6595 enrolled).
Additionally, adjustments for potential non-LDL-c factors that could have affected the risk of experiencing an event in this particular analysis were fairly paltry, not including non-HDL-c (omitting the apoB-containing lipoprotein, VLDL-c), HbA1c (only self-reported diabetes was included), and alcohol intake, to name a few. Finally, the magnitude of LDL-c-lowering for which a change in outcome risk was calculated was fairly small, with an absolute lowering of 20 mg/dL. The point estimate leaned in a direction suggestive of a small positive effect on reducing CHD risk (~7% RR reduction), but with wide confidence intervals (19% reduction to 8% increase).
Worth pointing out though, is that this point estimate is in a range compatible with Silverman et al.’s far higher-powered meta-analysis and meta-regression (to be discussed shortly), which found a significant relative CVD risk reduction of around 23% per 1 mmol/L (just under 39 mg/dL) decrease in LDL-c. Silverman et al.’s publication would suggest a 20 mg/dL reduction in LDL-c would result in about an 11.5% decrease in CVD risk, well within the confidence intervals of the RR in the WOSCOPS trial. As such, although a few major confounders plagued this particular trial, the results were still suggestive of an LDL-cVD relationship that is consistent with the findings from the reviews Ravnskov et al. claim they oppose.
The second study that Ravnskov et al. posit to proclaim no exposure-response relationship between LDL-c and CVD is fairly similar to the first28. It covered the Cholesterol and Recurrent Events (CARE) trial, an RCT carried out on 4159 patients with a recent MI, with a mean LDL-c and 139 mg/dL and 59 years, respectively. The main goal was to determine the effect of pravastatin treatment on coronary death or MI over 5 years. Once again, this trial demonstrated that after multivariate adjustment, LDL-c reduction significantly decreased the rate of the primary endpoints. However, it was in the studies secondary analysis that the authors discovered the rates of the primary endpoint in the total cohort only decreased progressively from the 10th (162 mg/dL) to the sixth (121 mg/dL) decile of LDL-c, beyond which no further reductions in outcome risks were observed.
They also determined that the absolute LDL-c reduction (as a numerical value or a percentage) was not a significant predictor of CVD mortality/morbidity risk reduction.
On these points, it is apparent this particular analysis mirrors that of the WOSCOPS trial. The authors used a different absolute LDL-c reduction in their risk reduction calculation, and their observations were prone to potential confounding resulting from variations in the baseline and final LDL-c concentration, among other CVD risk factors, within this non-randomized subgroup. Furthermore, only a single value for the point estimate of the risk ratio was given for the effect attributed to an absolute 25 mg/dL LDL-c change. Using this single value alone prevents consideration of whether or not the confidence intervals encompass the value that would be expected based on more extensive analyses evaluating the benefits attributed to greater reductions in LDL-c. As for the finding that beyond 121 mg/dL LDL-c led to no additional reduction in CVD risk in the entire cohort, a few things need to be clarified. First, in the analysis observing the influence of achieved LDL-c on CVD risk in the pravastatin group, there seemed to be a suggestive trend towards greater decreases with larger LDL-c reductions, though it was non-significant.
Alas, the detection of a linear relationship would likely be impaired due to the small number of cases in lower deciles of LDL-c reduction. The lack of cases for data analysis would undoubtedly limit statistical power and decrease precision, which is reflected by the large range of values encompassed in the confidence intervals for each decile. Such a result only emphasizes the need for larger samples with higher incidence rates in order to improve statistical power and reduce effect uncertainty. Also worth mentioning, as the study authors rightfully do themselves, is that this was a secondary prevention trial (post-disease), not a primary prevention trial like WOSCOPS. Secondary prevention trials indicate that the extent of plaque buildup is already large, meaning secondary prevention patients require more aggressive LDL-c reduction in order to have a better chance of responding to treatment than if it were initiated earlier in the disease process, given the cumulative and temporal nature of CVD. All of these factors must be considered when interpreting the study results as they clearly do not preclude the existence of an exposure-response relationship between LDL-c and CVD, as otherwise suggested by Ravnskov et al. who failed to consider other factors.
Finally, the last study referenced in support of the claim that a “true exposure-response” relationship between LDL-c and CVD hasn’t been established by statin trials is inappropriate. It’s again comical that it was even brought up as a part of a supporting argument. Alluringly titled “MIRACL”, this study was an RCT that assigned just over 3000 individuals to either placebo or atorvastatin soon after diagnosis of unstable angina or non-Q-wave acute MI29. These patients were then followed up for a mere 16 weeks to determine if atorvastatin treatment produced a reduction in the rates of death or non-fatal ischemic events. Shockingly, even in this extremely short follow up period, the results suggested those in the intervention group had a significant, 16% lower relative risk (2.6% absolute risk) of recurrent ischemic events, driven mainly by a reduction in symptomatic ischemic events requiring rehospitalization.
To the contrary, there were no significant differences in the rates of death, nonfatal MI, or resuscitated cardiac arrest.
This latter is totally expected, though, as it would be nothing short of an actual “MIRACL” for short-term statin treatment to produce a significant difference in rates of these outcomes in the context of secondary prevention. Accordingly, the authors very clearly stated, “This study was not powered to detect differences between treatment groups in the individual components of the primary composite endpoint”. Yet again this information was conveniently ignored and excluded by Ravnskovn et al.
In summary, not only is it unclear what Ravnskov et al. even mean by a “true” exposure-response relationship between LDL-c and CVD, the three references they used to demonstrate that this relationship does not exist include secondary analyses of previously conducted trials, which were extremely short, underpowered, and/or included a very specific sample demographic. All of these factors diminish the ability to even test causality with a modicum of certainty. There is simply no reason to seriously consider these studies when analysing the causality of LDL-c in CVD, and they most definitely do not refute the findings of the numerous meta-analyses of larger, higher-powered, better-controlled, and longer RCTs. Clearly Ravnskov et al. think otherwise, as they confidently declare these few trials provide a “strong argument against causality”, and continue to offer criticisms of said meta-analyses that are frankly weak, misleading, and borderline nonsensical.
For example, in the following paragraph, Ravnskov et al. turn their focus towards the meta-analysis and meta-regression carried out by Silverman et al. to investigate the influence of LDL-c-lowering via multiple methods on CVD risk30. Initially, Ravnskov et al. attack the study because the authors used reductions in major vascular events (MVE) as a measure of treatment benefit, saying that since this is supposedly defined differently in various other trials, it is “of dubious value”. While this is a great point to take into consideration when carrying out meta-analyses, it just doesn’t matter for their specific argument here. Silverman et al. made a concerted effort to match the inclusion trial outcomes into their analyses of MVE to the best of their ability. As made clear in the Silverman et al. supplementary data, the greatest extent to which the number of outcomes observed in the included trials differed was two or three at the very most, all of which were pre-specified in their inclusion criteria.
Moving onward, Ravnskov et al. make another absurd attempt to degrade the quality of the Silverman et al. analysis by claiming that using relative risk reduction is “highly misleading”. They state that in a case where only 2 patients out of 100 in the intervention group and only 1 out of 100 in the control group die, this would yield a relative risk reduction of 50%, but only an absolute risk reduction of 1%. Such a statement is astonishingly vapid and is ironically misleading itself. Silverman et al. are very forward in stating the following in their exclusion criteria, “(2) fewer than 50 clinical events during the course of the trial (to exclude small trials with unreliable hazard ratios)”, indicating their analysis considered the potential for a small number of cases to influence the risk/hazard ratios radically. Despite what Ravnskov et al. declare, it is completely appropriate to choose relative risk instead of absolute risk to gauge the efficacy of various interventions. Silverman et al. expand on why using only absolute risks is a problem, noting, “Second, the absolute risk reduction observed in a given trial will depend in part on the patient population, the endpoint, and the duration of follow-up; therefore, the analysis was of relative rather than absolute risk. As noted above, clinicians would need to integrate the estimated RR reductions from these analyses with a patient’s baseline risk to estimate the anticipated absolute risk reduction. Several tools are available for estimating patient risk.” Despite these points, Ravnskov et al. continue to trumpet that the “preferred way” to measure therapeutic benefits of statins would be to compare the absolute risk reduction per year of CVD, CHD, and total mortality. They provide nothing to support this assertion, assuming the reader will just take their word for it. Ravnskov et al. also don’t clearly explain why they only chose to focus on mortality outcomes, which can be problematic for numerous reasons, mainly the fact that mortality metrics are much less responsive to treatment in shorter trials (of which there were many), so this dilutes any potential effects. As highlighted by The Cooper Center Longitudinal Study, individuals at a relatively low ten year CVD risk don’t begin to show observable changes in mortality rates according to differences in LDL-c for more than five years at the very least, and these changes only increase with longer follow-up31.
Henceforth, focusing only on mortality outcomes results in the increased likelihood an analysis would fail to pick up on a meaningful difference in CVD rates unless the durations of the trials included were very long. Likewise, limiting the outcome to mortality causes an ignorance of reductions in non-fatal event rates (which would still have massive positive impacts on a patient’s quality of life and medical costs), and further restricting the values to only correspond to the effect over a single year would neglect the cumulative nature of treatment benefits. Therefore, this decision is hasty and misguided, once again making Ravnskov et al. the real victim of their own criticisms.
After making these statements, Ravnskov et al. proceed to explain they have carried out their own analysis to ascertain the absolute risk reductions in these outcomes. They included 11 trials in their analysis and claimed these were “ignored” by Silverman et al.’s meta-analysis, which is a misrepresentation. These trials were not included by Silverman et al. because they were captured in the analysis’ exclusion criteria, very plainly laid out in the methods section as follows: “Trials were excluded for the following reasons: (1) duration of less than 6 months (a timeframe during which a clinical benefit of lipid-lowering therapy would not be expected to emerge10); (2) fewer than 50 clinical events during the course of the trial (to exclude small trials with unreliable hazard ratios); (3) study population focused on participants with significant competing risks (ie, heart failure or chronic kidney disease because lipid-lowering therapy has been shown to be less clinically effective due to competing nonatherosclerotic risks6); or (4) experimental intervention with known off-target adverse effects on cardiovascular outcomes (which would impair the ability to judge the benefit of the LDL-c reduction).” Therefore, it should be apparent to Ravnskov et al. that, considering the potential for the types of trials to confound the results of meta-analysis, excluding them was the correct course of action. Nonetheless, Ravnskov et al. make absolutely no mention of this, gleefully carrying on by including the excluded trials in their rendition of the analysis. The results of this new analysis by Ravnskov et al. can be seen in their first and second figures. It shows that the “included” trials by Silverman et al. indicate a linear decrease in the yearly risk of CHD and all-cause mortality according to the difference in LDL-c between the control and intervention groups, but the “ignored” trials show an inverse or no relationship. Ravnskov et al. act as if this is somehow a meaningful finding despite knowing it would be expected considering the reasons provided by Silverman et al. for excluding these trials.
Funnily enough, even when inappropriately calculating the absolute risk reductions in only CHD/all-cause mortality per year, Ravnskov showed that LDL-c-lowering correlated with reductions in risk in the (likely) least confounded trials. This adds to the finding of Silverman et al. showing how a 1 mmol/L (38.7 mg/dL) reduction in LDL-c could influence the risk of hard cardiovascular events among groups at varying risks of CVD (as these types of calculations should be done). Silverman et al. remarked, “For example, a patient without known atherosclerotic cardiovascular disease who has a predicted risk of major vascular events of 15% and of hard cardiovascular events (cardiovascular death, MI, or stroke) of 10% within the next 10 years, lowering LDL-c level by 1 mmol/L (38.7 mg/dL) would be expected to result in absolute risk reductions of approximately 3.5% and 2.3%, respectively. A patient with known atherosclerotic cardiovascular disease who has a predicted risk of major vascular events of 45% and of hard cardiovascular events of 30% within the next 10 years, lowering LDL-c level by 1 mmol/L would be expected to result in absolute risk reductions of approximately 10% and 7%, respectively.” So, considering the temporal aspect of CVD, these 10-year risk differences would become more relevant and the results obtained by Silverman et al. are far from negligible, especially in individuals at higher CVD risk. As such, the so-called problems with this publication identified by Ravnskov et al. are not serious problems, and they are all considered by the authors and the study methodologies.
And as if they hadn’t showcased their deceptive behavior enough, Ravnskov et al. continue on to make similar attempts to refute certain points in Ference et al.’s consensus statement. They begin by noting that Ference et al. state the most compelling evidence for causality is “the presence of more than 30 randomized cholesterol-lowering trials that consistently demonstrate that reducing LDL-c reduces the risk of CVD events proportional to the absolute reduction in LDL-c”. Ravnskov et al. immediately object to this statement and claim “this is not true-exposure response”. Again, the “true” phrase is just as vague and confusing as the first time they mentioned it, and they don’t expand on what exactly it is supposed to mean. Even more astounding, Ravnskov et al. say that figure 5(a) in the Ference et al. paper, which shows the linear association, only contains data from 12 of the 30 supposed trials. This is nothing but an unabashed lie. Figure 5(a) demonstrates a strong, dose-response association between achieved LDL-c and five year CHD event rates found in a collection of 26 primary and secondary CVD prevention trials, as stated in the text.
The only reason over 30 trials are not included here is because this analysis didn’t contain trials that were not specifically secondary or primary prevention. Some included groups with and without previous history of CVD. But while the Ravnskov et al. remark does not accurately reflect the contents of Figure 5(a), it does reflect those of Figure 5(b), which includes 12 trials. However, that is only because Figure 5(b) instead displays results from trials observing the association between achieved LDL-c and percent atheroma volume change. As an astute reader may recall, Ravnskov et al. earlier stated that a relationship between LDL-c and atheroma volume does not exist, and the supporting evidence is only low quality collections of autopsy case-reports with no adjustments for confounders. The data from the RCTs here show this is not the case at all, exposing the extraordinary extent of Ravnskov et al. dishonesty and willful ignorance of evidence. It’s not even as if this is the only supporting evidence for the relationship anyway. A recent meta-analysis conducted by Li et al. depicted a dose-response regression of coronary artery plaque coinciding with the extent of LDL-c reduction, with serum LDL-c under 70 mg/dL eliciting the strongest effect32.
Following their fallacious comment about Figure 5, they continue on to say if all the trials included in their Table 1 were used, there would be no association between LDL-c-lowering and the 5-year absolute risk reduction of CVD. This is the exact same trick they tried to pull with Silverman et al. They included ostensibly “ignored” trials that were actually excluded rightfully due to the presence of known competing risks, short trial duration, low statistical power, and concomitant therapies that interact negatively with statins. It’s no surprise the association is diminished when these trials are included, making it devious of Ravnskov et al. to keep these details hidden. Curiously, they did choose to take one valid exclusion criteria into consideration, as they commented, “Ference et al. [3] claim that short-term follow-up (2 years or less) may be unable to demonstrate an association. We have, therefore, calculated the regression coefficients after having excluded such trials, but they do not differ much (included trials: r = +2.59 vs. +3.39; excluded trials: r = −0.1 vs. +0.15).”
But as highlighted earlier, trials under five years will most likely not be able to detect effects on mortality outcomes, making a defined cutoff of two years far from sufficient. Second, just by eliminating the few studies hindered by a shorter trial period it becomes clear that the slope of both included and excluded trials shifts upward, indicating duration is indeed an important variable to consider. Even more telling of how sloppy their attempts to discredit Ference et al. are is the fact that their figure (3) including these “ignored” trials says, “in 12 trials included in Table 4A in the article by Ference et al.”, yet there is no Table or Figure 4A in their article.
(Figure 4 and Table 4 from Ference et al. 2017)
Consistent with their previous jabs at many of the other analyses and arguments within the EAS’ consensus statement, the ones offered here are just as unavailing, representing a false portrayal of the actual quality of the research.
Section 6.2
To start this section, Ravnskov et al. assert the benefits of statin treatment are “exaggerated” by the authors of many articles referenced in the EAS consensus statement. First, Ravnskov et al. take issue with Collins et al.’s statin review because of the use of relative risk reduction values according to the degree of LDL-c-lowering33. Ravnskov et al. point out that the calculated 45% relative risk reduction in MVE per year with a 2 mmol/L decrease in LDL-c (from Collins et al.’s review) was obtained using data from the Cholesterol Treatment Trialists’ (CTT) meta-analysis, and remark, “according to Figures 3 and 4 in that article, the ARR of MVE was only 0.8% (1% for men and 0.2% for women), and the ARR of total mortality was 0.4% (both sexes).34”
However, the values obtained in the CTT publication are pertinent to a collection of sample populations different than the hypothetical Collins et al. calculated their values for; one with a much higher overall CVD risk. The difference in sample populations would change the values obtained from the absolute risk reduction calculations given that the baseline risk and subsequent change in event rates of CVD would differ, making the point by Ravnskov et al. based entirely on a fundamental misunderstanding of what the calculations were based on.
Next, Ravnskov et al. loop back to the Silverman et al. article, claiming that it suggested lower risk of MVE in primary and secondary prevention trials with only a 0.35 and 1.0% per mmol/L reduction of LDL-c per year, respectively. These numbers don’t come from the Silverman et al. article, nor were they mentioned previously by Ravnskov et al., so it is unknown where they originate. Nonetheless, it is repeated that 11 trials were ignored by Silverman et al. We have already clarified this was not the case; however, this time around Ravnskov et al. acknowledge that one of the exclusion criteria (total number of events lower than 50) didn’t apply to the “ignored” trials. I’m not entirely sure why Ravnskov et al. made this point, because there were clearly other exclusion criteria which the “ignored” trials were captured by. In one of the most embarrassing blunders of their entire article, Ravnskov et al. then carry on to say, “neither Collins et al. [1] nor Silverman et al. [2] mentioned that in four statin trials, where a high-degree lowering of LDL-c was compared with a low-degree lowering, no significant difference with respect to the number of MVEs was obtained, although LDL-c was lowered by 0.4–1 mmol/L more in the high-dose groups [53,55,56,61].”
As seen above, the four trials in question were carried out by Cannon et al., Koren et al., LaRosa et al., and Dujovne et al35-38. In direct contrast with the claims of Ravnsjov et al. the first three trials demonstrate that more intensive LDL-c-lowering significantly reduced MVEs to a greater degree than less-intensive LDL-c-lowering. The fourth trial was designed to assess the safety and lipid-lowering efficacy of Lovastatin, not its influence on MVE.
(Cannon et al. 2004)
(Koren et al. 2004)
(LaRosa et al. 2005)
(Dujovne et al. 1991)
These mistakes are pretty telling and highlight the perfunctory nature of their attempted criticisms.
Finally, in the closing sentences of this section, Ravnskov et al. claim that the “most important outcome”, an increase in life expectancy, has not been mentioned in any of the cholesterol-lowering trials. They then turn their attention to a recent calculation in an article published by Kristensen et al. that showed the increase in life expectancy seen with statin treatment was no more than an average of a few days39.
As mentioned previously, it’s arguable that death is the most important metric to use when assessing the efficacy of lipid-lowering therapy. Death ignores effects on events and quality of life, and is much less sensitive an endpoint which makes it harder to pick up on meaningful effects unless the trials being used are of a long duration and possess high statistical power (not the case for many of those used here – half of them weren’t even 5 years long). These points aside, the claim about life expectancy using the Kristenson et al. analysis still remains a misleading one that overlooks other factors capable of influencing the observed effect, not to mention errors in the analysis itself that produce erroneous values.
The first thing to note, besides study duration, is that multiple trials in the Kristenson et al. analysis had a sample population with numerous health issues that would skew the statin efficacy for lowering CVD risk, namely heart failure and chronic kidney disease. Furthermore, a few of the trials had a sample population with relatively low LDL-c levels to begin with, which would shift the group’s overall CVD risk and decrease the subsequent increase in lifespan seen with additional lipid-lowering. Another issue to add to these is that only all-cause mortality was considered. Although CVD is one of the leading causes of death, there are still numerous non-CVD causes of death that would not be affected by statins at all, thus blunting the survival extension values that would be conferred via reduced CVD mortality. Lastly, calculating the average survival duration precludes the potential for detecting a non-uniform distribution of the benefit. For example, if there was a large proportion of people who experience a very long life extension and a small proportion that experience small or negligible life extension (or vice-versa), a simple mean survival duration value would not be truly representative of the potential effect. The study authors discussed many of these caveats in the last few paragraphs of their publication, demonstrating they clearly acknowledge these issues might be altering the results. Again, Ravnskov et al. neglect to bring attention to these critical points.
Luckily, just over five years ago Finegold et al. carried out an analysis that took the majority of these error sources into consideration and sought to identify how the lifespan gain from primary prevention statin treatment may be distributed differently among various populations40. Not only did they find that primary prevention with statin treatment was equivalent to that shown to reduce CVD mortality by 30% in a previous meta-analysis, but also that statin treatment coincided with a much larger mean life extension for men aged 50 years old with average CVD risk (7 months versus 5-19 days in Kristense et al.’s analysis). They discovered a large disparity in the distribution of said extension and provide the example that for the previously described group of men, 93% saw no appreciable gain in lifespan, but 7% saw an astounding mean of 99 months of life gained. That is over 8 years.
Furthermore, the distribution of life extension was largely modified by time of therapy initiation and baseline CVD risk of the demographic. With earlier initiation of statin treatment and higher baseline CVD risk, exactly the population such a medication would be most indicated in, a larger proportion would see an even greater mean extension of life.
Henceforth, the values for life expectancy with statin therapy (in the right conditions) could be up to hundreds of times greater than suggested by the research cited by Ravnskov et al. It can even be argued that, even in the best quality analysis available, many unaccounted variables could lead to an even greater benefit than detected. Such variables include, but are not limited to: other factors besides elevated LDL-c (glycemic control, overweight/obesity, hypertriglyceridemia, diet, alcohol consumption, etc.), the potential that average ages of mortality will increase from the values used for the calculations in Finegold et al.’s analysis (which would cause their values to be underestimates), and the infeasibility of accounting for all other causes of death unrelated to CVD.
So, in conclusion, only considering mortality metrics entails ignorance of other parameters of quality of life, and there is a gross underestimate of life extension in the analysis Ravnskov et al. cites. The uneven distribution of benefit (favoring those who would require statin therapy even more) and the chance that the values from higher-quality research may fall short of the actual benefit of statin therapy indicates that the only misleading claims are those from Ravnskov et al.
Section 6.3
In this brief section, Ravnskov et al. remark that after the adoption of New Clinical Trial Regulations (specifying trial data must be made public) in 2005, “benefits from statin trials have virtually disappeared”, referring to their figures 4 and 5. These figures cover the absolute CHD and total mortality risk reductions seen in the trials from Silverman et al.’s in addition to the “ignored”, or rightfully excluded, trials.
Once again, this is not only highly uncharitable, but just plain incorrect. When looking over their figures for Silverman et al.’s analysis, only 6 and 8 trials after 2005 are considered for CHD and total mortality, of which 4 trials for each outcome were not included in the original meta-analysis on the basis of falling under their exclusion criteria. This is nowhere near enough trials to make a claim about the efficacy of statins on mortality outcomes, and it is disingenuous considering that most of the trials are heavily confounded to the point of warranting exclusion. Multiple trials since implementation of the new regulations have shown benefit. For example, a later meta-analysis by Yebyo et al. included a multitude of these trials and they did not meaningfully alter the significant effects on MVE or all-cause mortality, making this just another instance where Ravnskov et al. fail to substantiate an overstated claim41.
Section 6.4
Next, the discussion shifts focus to adverse effects associated with the use of statins, starting with a comment asserting that Collins et al. may be incorrect in saying that adverse effects are infrequent and that RCTs are the best tool to assess their frequency. This assertion is founded on the notion that “many drug-related adverse effects in other therapy areas have only emerged from observational studies and post-marketing surveillance”, in addition to the chance for a run-in period to “weed out” those who suffered adverse effects and did not want to continue therapy. The first comment is worth considering; however, observational research should not be the main basis for determining adverse effects given the high likelihood for nocebo effects to manifest in the absence of a control group receiving a placebo to compare to. The second point is largely irrelevant, as in a patient setting those who experience immediate side effects upon treatment initiation would discontinue therapy and seek an alternative option. Regardless, in the following sentence Ravnskov et al. point to two trials that supposedly possess no run-in period and where serious side effects were recorded in an appreciable number of participants receiving statin therapy. They state, “in SAGE [64], serious side effects were recorded in more than 20% in both groups, and in IDEAL [55], the number was almost 50%.”
Once more, showcasing the lack of rigor in the work of Ravnskov et al., neither of these abbreviations match the names of the trials they linked in their references. The first, supposedly SAGE, was actually an RCT on the use of Rosuvastatin in older patients with elevated LDL-c and systolic heart failure. This population would typically not be indicated for treatment due to the competing risk of heart failure42. To make things funnier, this trial did use a run-in period (though only with a placebo to determine potentially poor therapy compliars), and throughout the trial those in the treatment group had less serious side effects than those in the control group. This doesn’t support the original claim made by Ravnskov et al.
The second reference Ravnskov et al. linked was to the ALLIANCE study, not IDEAL. ALLIANCE was an open-label, prospective, randomized trial comparing the effect of atorvastatin to “usual care” (chosen by the patient’s health provider)43. This trial was actually discussed earlier in the Ravnskov et al. article since they also cited it in support of their incorrect claim that more intensive LDL-c lowering did not elicit further CVE risk reduction. Throughout the trial, serious adverse events occurred in 40% and 42% of the atorvastatin and usual care group, again indicating that treatment did not induce a meaningfully different rate of adverse events, instilling more confusion as to why Ravnskov et al. even included it in their references.
In all fairness, since the names of these trials didn’t match up with the ones they were discussing, the charitable thing to do would be to observe the study results of what they meant to cite. The first study (SAGE) was “a 12-month, prospective, international, multicenter, randomized, double-blind, double-dummy, parallel-arm trial that compared the effect of atorvastatin 80 mg/d with that of pravastatin 40 mg/d on the total duration of myocardial ischemia as assessed by 48-hour ambulatory ECG” on an elderly sample (mean age about 73) with history of CHD44. One thing that stands out right away is that this study lacked a placebo group, preventing the investigators from identifying whether any adverse events resulted from a placebo/nocebo effect. Regardless, while serious adverse effects were seen in “more than 20% in both groups” as Ravnskov et al. stated, they failed to mention that when CVD events were excluded, serious adverse events only occurred in 12.1% and 11.9% of participants in each group, respectively. These are hardly concerning rates of adverse events for a medication used in an elderly sample, and again, we cannot parse out any placebo/nocebo effects without the presence of a control group.
The other study (IDEAL) that Ravnskov et al. discussed was another prospective and randomized, but open-label and blinded end-point trial on patients aged 80 or younger with a previous MI. The study compared the efficacy of usual-dose simvastatin to high-dose atorvastatin, again with no placebo group45. The number of total serious adverse events reported were 2108 (47.4%) and 2064 (46.5%) in the simvastatin and atorvastatin groups, which upon first glance do appear to be fairly high. That being said, there were CVD events in 1370 (30.8%) and 1176 (26.5%) individuals in each group, and if these were included from the total events, that would cut the adverse events due to the treatment down to around 10-15% in each group. Further, only 186 (4.2%) and 426 (9.6%) of the adverse events warranted discontinuation of the treatment in each group, and no type of adverse event responsible for this discontinuation was present at a rate higher than 2.2%. So, similar to the previous trial, these numbers are not shocking at all. They do not suggest that adverse events are a major issue with statin treatment, contrary to the position held by Ravnskov et al.
Following the concerns about supposedly high adverse event rates in statin trials, Ravnskov et al. take issue with Collins et al.’s declaration that only 0.01% of individuals treated with statins contract myopathy. Ravnskov et al. object to this, noting that “in most statin trials, myopathy is only recorded if creatine kinase (CK) is more than 10 times higher than normal.” It’s unclear as to why Ravnskov et al. make this objection, given that elevated CK (in addition to other physical symptoms like muscle pain/fatigue) are part of the diagnostic process for myopathy, and are also used to inform professionals how to approach medical management46.
Instead of accepting these methods, Ravnskov et al. decry this decision and claim, “However, in a study by Phillips et al. [73], microscopic examinations of muscle biopsies from statin-treated patients with muscular symptoms and normal creatine kinase levels showed signs of myopathy. When patients stopped treatment, their symptoms disappeared, and repeated biopsies showed resolution of the pathological changes.” The study they mention by Phillips et al. is a case study of four, though. Yes, just four people47. These individuals were originally part of a much larger statin trial and experienced myopathy-like symptoms to a magnitude great enough to warrant discontinuation of the statins, despite not exceeding CK levels greater than ten times normal. They were not forced to continue statin treatment, and the symptoms and signs of damage were reversible with cessation/alteration of treatment.
What Ravnskov et al. is trying to assert with this reference is up for question, because as eloquently stated in a response from Scott Grundy, “For high-risk persons, the proven efficacy for preventing cardiovascular disease outweighs the unlikely possibility of permanent muscle damage48. Phillips and colleagues’ preliminary results certainly do not provide adequate information on the spectrum, scope, or prognosis of myopathy with normal creatine kinase levels during statin therapy. For these reasons, the prescription of statins for eligible patients should continue despite the current results. Moreover, before discontinuing therapy, physicians should carefully evaluate any patient receiving statins who report muscle symptoms. In most cases, the symptoms will be found not to be consistent with chronic myopathy, and often they will not be related temporally to statin treatment. High-risk patients, in particular, should not be deprived of major cardiovascular risk reduction just because they display symptoms not clearly documented to be closely related to statin therapy.” Therefore, while the Phillips et al. findings give insight into a potential group that may require closer attention, they say little about statins’ overall benefit and safety profile.
Continuing with their commentary, Ravnskov et al. further state, “To reject the frequent occurrence of muscular problems with the argument that muscle symptoms are nocebo effects is also invalid. In a study of 22 statin-treated professional athletes [74], the authors reported that 17 (77%) of the athletes terminated treatment because of muscular symptoms, which disappeared a few days or weeks after drug withdrawal.” The fact they mention this publication as if it supports their claim is just bizarre. It’s just another case study of a small subset of individuals who had difficulty tolerating certain statins49. It’s a mystery where Ravnskov et al. got the impression anyone was claiming that the (very low) frequency of muscular problems is entirely due to nocebo effects. This is especially funny considering there was no placebo or control group in this study, and therefore no ability to assess the possibility. Likewise, the argument that muscular symptoms are nocebo “is invalid” is highly dismissive, and Ravnskov et al. don’t explain why this would be true. In reality, according to recent research, it seems far more likely that an appreciable proportion of muscle-related symptoms attributed to statin use actually are nocebo. Last year, a cleverly designed double-blind, three-group, n-of-1 trial carried out by Wood et al. shed some light on this previously controversial hypothesis. This study recruited a group of patients who had previously discontinued statin therapy due to suspected side effects, and assigned them to statin tablets, placebo tablets, or no tablets (for a total of 4 months taking each) in a random sequence for a year. During this period, patients recorded daily symptom severity scores. Following completion of the trial, it was found that average monthly symptom intensity ratings did not differ significantly between months where subjects took statin or placebo tablets, but that both corresponded with significantly higher symptom intensity ratings when compared to no tablet months. Correspondingly, the authors concluded that an astounding 90% of the symptom burden of a statin challenge was also seen with placebo, clearly suggesting a widespread nocebo effect50. Thanks to these results, half of the patients had successfully restarted statins 6 months after the completion of the trial, four others planned to, one could not be contacted, and the others opted out for various reasons.
So, in addition to the fact it appears perfectly reasonable to expect nocebo effects may comprise a non-negligible proportion of subjective statin-related side effects such as myopathy, nothing Ravnskov et al. bring to the table changes the validity of Collins et al. statements about the frequency of this type of side effect. The entire thing is a complete non-sequitur. The information that Ravnskov et al. share here just indicates that there may be certain subsets of the population who experience rarer, novel side-effects and therefore require different treatments for elevated LDL-c. None of the authors of these publications deny this, and it doesn’t disprove anything previously stated. It only seems as though this was another concerted effort by Ravnskov et al. to inappropriately dissuade individuals from starting a statin if one is advised.
In the next few paragraphs, Ravnskov et al. deploy extreme shotgun argumentation, dumping a host of links to cross-sectional and case-control studies demonstrating associations between statin use and various adverse outcomes. They state, “Furthermore, case–control and cross-sectional studies have shown that statin use is observed significantly more often among patients with cataracts [76], hearing loss [77], suicidal ideation [78], peripheral neuropathy [79], depression [80], Parkinson’s disease [81], interstitial cystitis [82], herpes zoster [83], impotency [84], cognitive impairments [85–88], and diabetes [89,90]. In some of these studies, the side effects disappeared with discontinuation of the statins and worsened with rechallenge [74,84,85].” Interestingly, they do not repeat the “correlation does not equal causation” mantra they constantly deployed against LDL-c and CVD, and fail to mention the small sample sizes, under-adjustment for confounders, large number needed to harm values, and far higher quality investigations overturning many of the findings of the studies they linked. For example, a large meta-analysis of RCTs showed that statins do not induce depression symptoms in those without depression at baseline and may even elicit small improvements in symptom scores for those clinically depressed at the beginning of treatment51.
Meta-analyses of both observational studies and RCTs also demonstrate that statin use decreases or has no effect on the incidence of all-cause dementia, Alzheimer’s disease, and mild cognitive impairment among both normal and cognitively-impaired participants52-54.
(Chu et al. 2018)
(Chu et al. 2018)
(Chu et al. 2018)
(Poly et al. 2020)
(Poly et al. 2020)
(Ott et al. 2015)
(Ott et al. 2015)
A systematic review and meta-analysis of cohorts, case-control studies, and RCTs concluded that statins do not significantly increase the risk of cataracts. Despite cohorts hinting there may be an increased risk, neither case-controls nor RCTs support this, and sensitivity analyses indicate that the effect in the cohorts was confined to studies with shorter follow ups55.
Another meta-analysis of 8 case-control studies and 9 cohort studies determined that statins may decrease, not increase, the risk of Parkinson’s Disease56.
A recent meta-analysis of prospective and case-control cohorts revealed that statin exposure likely does not increase the risk of peripheral neuropathy, with the highest (and frankly obscene) RR coming from a completely unadjusted analysis57.
Meta-analyses of RCTs have suggested that statins do not increase the risk of developing erectile dysfunction (ED), and they may even improve erectile function among those with signs of ED at baseline58-60.
(Elgendy et al. 2018)
(Cai et al. 2014)
(Cai et al. 2014)
(Kostis et al. 2014)
To Ravnskov et al.’s benefit, one meta-analysis of cohorts did suggest statins may slightly increase the risk of herpes zoster, although the authors were not confident in the results because they varied substantially by study region and were characterized by a high degree of heterogeneity61.
Finally, according to a network meta-analysis of RCTs, high doses of certain statins such as atorvastatin and rosuvastatin may actually increase the risk of diabetes, which does indicate this is also something worth considering when prescribing them62. It may be the case that certain statins that are not linked to higher diabetes risk should be prescribed for pre-diabetic or at-risk patients to best avoid issues here.
Overall, the available meta-analyses, with far more rigorous methodology and much larger samples, show that almost all of the harms Ravnskov et al. fervently assert are attributed to statins are completely false. Many suggest the effect might be in the opposite direction. While the incidence of herpes zoster and diabetes may indeed increase with statin use, the effects are small, likely specific to certain statins, and are characterized by a large amount of uncertainty. They clearly don’t come close to the magnitude and extent of the positive effects observed with statin use for whom they are indicated. Collins et al. themselves directly echo this sentiment in their review, stating that new-onset diabetes is a known potential side effect, but that the clinical relevance is questionable considering the disparity between the small risk of this side effect and the large benefit of statins in primary CVD prevention.
Immediately following this egregious wave of misinformation, Ravnskov et al. close off the paragraph with another sentence riddled with false conclusions, this time based on mechanistic speculations that do not correspond with a large body of human outcome-based evidence. They declare, “As cholesterol is a vital substance for the renewal of all cells, and since statins also block the production of other molecules necessary for normal cell function [75], it is not surprising that statin treatment may result in side effects from many different organs.” But plausible this may sound, it’s just not true. The vast majority of cells have the ability to produce cholesterol as needed. Plus, if cholesterol synthesis is insufficient, cells can increase LDL-c receptor expression to increase cholesterol uptake from circulation63. This is actually one of the primary mechanisms by which statins decrease plasma LDL-c.
Therefore, statin use does not induce any sort of cholesterol “deficiency” as Ravnskov et al. seem to imply, at least outside of patients with underlying compensatory and regulatory cholesterol issues. In addition to this, research on numerous medical conditions and multiple meta-analyses of RCTs, prospective, and retrospective cohorts have demonstrated that sustained exposure to low levels of serum LDL-c is safe and produces pronounced reductions in CVD mortality, with very low potential for side effects, none of which have been reliably attributed to lower LDL-c directly64.
The only question remains whether or not extremely low LDL-c, as in under 15 mg/dL, may pose additional risks. The authors of the above review note that the current short term evidence suggests this is unlikely, though. As such, the claim that statins elicit harmful side effects via LDL-c reduction due to cholesterol’s vital role in cellular processes does not fit with reality, no matter what Ravnskov et al. suggest.
In the next paragraph, Ravnskov et al. argue against Collins et al.’s claim that statin treatment may protect against cancer, repeating their earlier suggestion that low cholesterol actually increases cancer risk. In an attempt to support this assertion, they cite the editorial by Ravnskov that contained RCTs with low incidence rates for a random collection of cancers, as well as nine cohorts, still continuing to ignore that they are directly contradicted by multiple, larger meta-analyses as covered in the discussion of Section 5.1. This time they take things a step further, remarking, “For instance, several experiments on rodents with lipid-lowering drugs produced cancer [91], and in nine human cohort studies, cancer rates were inversely associated with cholesterol levels measured in healthy people 10 to more than 30 years earlier [24]. Therefore, case–control studies in which the incidence of cancer in statin-treated patients was lower than in controls are invalid because many untreated individuals have low cholesterol, and those on statins have lived most of their lives with high cholesterol that may have provided protection from developing cancer.” To start things off, the fact they refer to animal-based data to support this claim is astonishing given that translation rates to humans, especially for carcinogenicity tested in rodents, are not reliable65.
The fact Ravnskov et al. actually believes these low-quality studies that fail to account for major cancer risk factors, while entirely ignoring or dismissing the massive body of evidence showing that lower cumulative LDL-c does not increase the risk of cancer, is utterly staggering. To top things off, they close out this section by stating that while the side effects mentioned previously may be small, the total number can be substantial when taken together, especially in patients treated for many years. It is difficult to perceive this as anything but an uncharitable conclusion that displays their bias against statins, since, as shown above, many of these side effects are not even reliably attributed to statin use (or their use actually decreases their occurrence). Statin use would not be continued for years if someone were experiencing major side effects, and there is not a modicum of consideration from Ravnskov et al. as to how cumulative effects may (and do) apply to the risk reductions observed for CVD endpoints. While the possibility of adverse effects is real, as with any medical treatment, the amount that have been determined to be caused by statin use, and the frequency at which they occur, are nowhere even close to what Ravnskov et al. wants the reader to believe.
Section 6.5
In the following section, Ravnskov et al. move on to question whether or not treatment with proprotein convertase subtilisin–kexin type 9 (PCSK-9) inhibitors, a new potent cholesterol-lowering drug, can “improve the outcome”. I assume they are talking about statins with respect to CVD mortality. They mention the FOURIER trial, which was the largest trial on this treatment to date, noting that after 2.2 years, there appeared to be a significant reduction in major vascular events in the group treated with the PCSK-9 inhibitor evolocumab66. However, Ravnskov et al. state that CVD mortality and total mortality increased, though not with statistical significance, and question why the trial was ended after only 2.2 years as if the sponsor had nefarious intent. Finally, they close off this section by stating this trial is, “yet another proof that there is no exposure-response between LDL-c and total or CVD mortality.” In reality, this is only further proof that they selectively report findings to favor their argument, offer uncharitable interpretations, and reach conclusions that don’t follow logically. It would be absurd to expect CVD or all-cause mortality rates to be significantly influenced in such a brief follow up period. In addition, their suggestion that these outcomes “increased” is highly disingenuous. None of these outcomes differed more than a tenth of a percent across groups. Given that the analysis was far from capable of confidently establishing an effect for either of them, it would be inappropriate to assume any differences that may exist were genuine. That being said, the incidence of the primary and secondary endpoints, comprised of more responsive outcomes (MI, stroke, hospitalization for angina, and coronary revascularization, even including CVD death), were significantly lower in the group treated with evolocumab, mostly driven by an impressive 27% reduction in MI.
As for the early cessation of the FOURIER trial, the reason for this is explicitly stated by the authors, who stated, “Although the median follow-up period in FOURIER was originally planned to be approximately 4 years, an event rate that was approximately 50% higher than had been postulated led to a shorter required duration of follow-up to accrue the prespecified number of events.” To continue the trial for years after observing such drastic reductions in CVD events would be unethical, considering it would require denying the control group of a treatment that would likely improve their health. So, in closing, to conclude the FOURIER trial was “proof” that no exposure-response relationship between LDL-c and CVD/total mortality exists is spurious, since the duration was too brief to assess the effect on these less-sensitive outcomes meaningfully. If anything, the results regarding more sensitive outcomes strongly indicate an exposure-response relationship, and that longer treatment would likely reveal the same relationship with the mortality outcomes that Ravnskov et al. chose to hyper-focus on. Consistent with this position, a recent Cochrane review suggested that results from trials using the PCSK-9 inhibitor alirocumab showed significantly decreased CVD, MI, and CVD mortality risk compared to placebo. Likewise, based on a smaller number of studies (only 3, including FOURIER), evolocumab significantly decreased CVD, MI, and stroke incidence, but not mortality. This again demonstrates the need for larger samples and longer follow ups to reveal an effect on the latter67.
Therefore, by selectively choosing a single trial, honing in on outcomes that were not appropriately designed to signal effects, and ignoring a large amount of robust contradicting data, Ravnskov et al.’s cursory discussion of PCSK-9 inhibitors does nothing but highlight their repeated lack of investigative rigor. In contrast to their conclusion, the results of larger analyses indicate a benefit of this therapy that conforms with an exposure-response relationship between LDL-c and CVD pertaining to both events and mortality.
Section 7.1
In the first few paragraphs of this section, Ravnskov et al. suggest that many observations conflict with the “assumption” that high LDL-c is the cause of increased CVD and premature death among individuals with familial hypercholesterolemia (FH). The first of these observations was documented in a publication focused on data from the Simon Broome Registry for FH68. This registry consisted of a sample of patients who were referred to the participating clinic due to the presence of xanthomas or a personal/family history of premature vascular disease. After initial records were collected, patients were followed for up to 9 years to observe patterns in CVD and all-cause mortality among different age groups. According to Ravnskov et al., results showed, “only a small percentage of FH individuals die at an early age, and the mortality among the elderly does not differ from the mortality of the general population despite their high LDL-c.” What they failed to mention was that, aside from the small and non-representative nature of the sample, CHD mortality rates among all age groups except the oldest (60-74 years old) were over 5 times greater for individuals with FH than the general population.
Furthermore, the individuals who were diagnosed with FH would have initiated lipid-lowering treatment. There is a strong chance this reduced the ‘actual’ FH mortality rate that would have been observed in the absence of lipid-lowering treatment. Even so, the number of follow-up years for those in the oldest group was only around one-tenth of the value for the entire sample, leaving this particular analysis remarkably underpowered to detect any potential outcome differences. As if these weren’t enough to demonstrate the unreliability of these observations, the potential for survivorship bias is incredibly high in this type of study. Survivorship bias has historically caused largely erroneous results to be produced in other health-related research69.
Regarding this study, it is possible that untreated or undiagnosed FH individuals deaths would have died at earlier ages and are therefore not included in this sample. This would dilute the true rate of CHD death in FH groups because the mortality rate in the lower age ranges is missing from the data. In addition, the resulting sample of older FH individuals would likely possess a greater number of lipid or non-lipid related characteristics that decrease their overall susceptibility to CVD events, given this group must have avoided the frequently observed high mortality rate seen in those with FH at younger ages. The study authors even expressed concern in the final few paragraphs, stating that care is required when interpreting their findings due to the selective nature of their sample. Oddly, the authors mention the chance for undiagnosed individuals to decrease CHD rates observed, yet don’t consider the opposite occurrence despite what I just discussed. Regardless, the authors clearly state that the disease rates were so high they would persist even if a large number of FH individuals who did not have “evidence of cardiovascular disease” were included. Again, none of this was acknowledged by Ravnskov et al.
The next study that Ravnskov et al. offers to support the notion that high LDL-c is not a cause of CVD morbidity and mortality was conducted by Mundal et al. These authors carried out a similar analysis of 4688 Norway FH residents aged between 0-92 years old70. The subject information was obtained from the Unit for Cardiac and Cardiovascular Genetics (UCCG) Registry, and the sample was followed for up to 19 years. The rates of CVD and non-CVD mortality during this period were compared to age-matched samples from the Norwegian Cause of Death Registry to determine any differences compared to the normal population. The only statement Ravnskov et al. make about this analysis is, “During that time, 113 died, whereas the expected number in the general population was 133. The mortality benefit cannot have been due to lipid-lowering treatment because there was no significant difference between the number on such treatment among those who died and those above the age of 18 years who survived (88.2% vs. 89.1%).” So, to start, Ravnskov et al. ignore the substantially greater rates of CVD mortality among FH groups in all the included age groups up to 70 years old, where they then become equivalent to that of the general population. This is essential information for Ravnskov et al. to avoid mentioning. Sure, there were no significant differences in the highest age group, but this isn’t groundbreaking when considering this is the age group where the highest incidence of CVD occurs in the general population. It means the FH group died younger.
Similar to the previous study, Ravnskov et al. also totally ignores the fact that the FH group in this age range likely has a propensity towards greater resistance to CVD due to survivorship/selection bias. There is no mortality “benefit” in the FH group. To suggest such would be entirely speculation and dismisses the existence of FH-related CVD deaths that would not be captured in the registry due to undiagnosed individuals. Again, this was directly noted by the study researchers, who said, “FH is both underdiagnosed and undertreated in the general population. After the Netherlands, Norway has the highest percentage of genetically diagnosed FH worldwide, given an estimated prevalence of 1 in 500; however, only 26% of persons with FH are genetically diagnosed in Norway, based on an estimated prevalence of 1 in 300 with heterozygous FH. Because so many with FH are undiagnosed, the true mortality rates might be different, and even higher SMRs for CVD mortality could be expected.” Therefore, the undiagnosed individuals could easily make up for the minor gap between observed and expected deaths. The ‘true’ observed mortality rate could likely even surpass the expected rate by a large margin. Likewise, the choice of Ravnskov et al. to only look at total all-cause mortality numbers without acknowledging the possibility that any discrepancies could be due to non-CVD related deaths occurring at a high frequency in the normal population (which they did) is devious.
As a side note, although not directly related to the argument that Ravnskov et al. were attempting to make using these results, it is comical that the authors note their analysis showed statins likely don’t increase cancer mortality. They describe that the overall cancer rate was lower in the FH group, in which most individuals were exposed to statins for over 20 years. That said, one cannot forget, according to Ravnskov et al., “correlation does not equal causation”…except when it’s convenient for them.
Lastly, the final comment by Ravnskov et al. about the mortality difference not being attributable to lipid-lowering treatment because there was no significant difference between the number of those who died on treatment and those who survived is also naive. It completely disregards the notable differences in the time at which statin treatment was initiated, the dose used, and an uneven distribution of longevity benefit, all of which would modulate the observed effect. So, in reality, although these two studies were proposed as “exceptions” to invalidate the LDL-cVD relationship ostensibly, not even the researchers are convinced this is the case. The lack of confidence from the study authors is evidenced by the fact they openly characterize multiple limitations and considerations that would explain the results and are consistent with the current LDL-c paradigm.
Section 7.2
In the subsequent section, Ravnskov et al. continue their discussion about FH, suggesting, “If high LDL-c causes premature CVD in FH, the LDL-c of those with CVD should be higher compared to others, but at least six studies of untreated FH individuals have shown no significant differences in LDL-c or age [95–100]. It has also been shown that FH relatives without FH may have shorter lives than the general population [101]. Most likely, a small subset of FH individuals and their relatives inherit CVD risk factors that are more important than high LDL-c on CVD outcomes71-76.” The argument that Ravnskov et al. present here is completely illogical. The idea that LDL-c should always be higher among those with CVD and that if it is not then it does not cause premature CVD essentially assumes that it is either the only risk factor, or outweighs everything else by a factor so large that even combining them all wouldn’t influence the risk to a greater degree. It is well known and widely accepted that other factors modify CVD risk to a substantial degree. Therefore, someone with FH that has a high LDL-c and multiple additional risk factors would be at a much higher risk of experiencing premature CVD than someone with FH without additional risk factors. This is exactly what the studies they cited demonstrate.
Before delving into the specifics, it is also important to note that in almost all of the studies, a diagnosis of CVD was based only on the presentation of angina alongside a positive exercise test, confirmed stenosis of >70% in a major vessel, a MI, and/or coronary artery bypass grafting. Henceforth, even those who likely had extensive plaque buildup, but hadn’t had an angiography performed, experienced an event, or suffered consistent symptoms yet, would be deemed as CVD free when chances are they are far from it. Ravnskov et al. don’t make much of an effort to clarify this, leaving the reader with the impression that those “without” CVD were in perfect health and free of risk when it is not the case. With that being covered, as mentioned, all these observations highlight is that when combining multiple factors that would compound CVD risk in someone with FH, said individual is more likely to experience CVD symptomatology, a major event, or have a significant blockage identified via angiography. This is not a novel finding, nor does it preclude the role of elevated LDL-c in CVD risk. As seen below, most of the groups diagnosed with CVD in each of the studies Ravnskov et al. cited had similar LDL-c compared to the CVD “free” group but contained substantial differences in one or more additional risk factors for CVD. These included older age, elevated Lp(a), higher rates of smoking, higher triglycerides, higher rates of hypertension and diabetes, higher BMI, and lower HDL.
(Seed et al. 1990)
(Seed et al. 1990)
(Wiklund et al. 1990)
(Vuorio et al. 1997)
(Wittekoek et al. 1999)
(Smilde et al. 2000)
(Cenarro et al. 2003)
Also worth mentioning, the few studies that directly measured atherosclerotic plaque progression showed that virtually all FH individuals had substantial buildup and intima-media thickening regardless of whether or not the subject was diagnosed with CVD. This emphasizes the importance of my earlier point that those denoted as CVD “free” indicates they haven’t yet experienced the symptomatic manifestation of the disease.
(Smilde et al. 2000)
(Smilde et al. 2000)
(Wittekoek et al. 1999)
Curiously, after making an initial statement that implied LDL-c must be higher in those with FH and CVD for it to be causal, Ravnskov et al. continue on to acknowledge the existence of other risk factors in the last sentence of this section. However, it is only to suggest that anyone who has FH and is diagnosed with CVD or experiences a major event probably ends up in that position due to the presence of “CVD risk factors that are more important than high LDL-c on CVD outcomes.” It’s difficult to perceive this as anything but a disturbing display of motivated reasoning. Instead of acknowledging the most likely answer–that high LDL-c can significantly increase CVD risk and additional lifestyle-related factors can compound this risk–they somehow decide that high LDL-c has little or no role in CVD risk in individuals with FH and that they must inherit other more important factors. As much as they try to warp the narrative in this direction, their citations don’t even support their attempts to do so. Their citations demonstrate that extensive plaque and increased intima-media thickness are observed in almost all FH subjects. Those with one or more risk factors alongside elevated LDL-c are naturally the first to be diagnosed with CVD or experience a major event. Try as they may, in no way does this eliminate LDL-c as a causal factor in CVD development among individuals with FH.
Section 8
In the penultimate section, Ravnskov et al. turn their attention towards changes in CVD mortality across the world within the past few decades. They say that the presumed reason CVD mortality has decreased is the increased use of statins, but that this is “highly questionable”. They don’t specify anyone that has actually said this, nor do they entertain the possibility (that most health professionals agree with) that it could be partly statins, partly other lifestyle modifications, and several other factors. This leaves me wondering why they chose to go after such an assertion at all. Nonetheless, they continue on to say, “In a Swedish study including 289 of the 290 municipalities, no association was found between statin use and the change in mortality from AMI (Acute Myocardial Infarction) [102].” The study they link here is an ecological study that sought to determine if the increased use of statins in Sweden’s municipalities between 1998 and 2002 influenced AMI mortality from 2000 to 200277. The author’s analysis showed that with increased statin usage, the incidence of AMI decreased slightly for men, but not as notably for women. In contrast, no correlation of statin use with AMI mortality was observed after adjustment for socio-economic deprivation, anti-diabetic drugs, and geographic coordinates.
Considering the relatively low baseline rates of CVD incidence/mortality in the observed population, the lack of adjustment for numerous risk modulating variables, and an inability to assess statin adherence, mean these results are as expected. Likewise, the probability that a two to four-year follow-up would reveal notable changes in the already relatively low mortality rates of a disease that takes decades to progress would be obscenely low, covered ad nauseum at this point. If anything, Ravnskov et al.’s repeated citation of short-term analyses with a “lack of effect”, similar to this one, emphasize the importance of considering the temporal, exposure-response nature of the LDL-cVD mortality relationship that Ravnskov et al. adamantly claim does not exist. Taking all of this into account, using this analysis as a basis for drawing any conclusion about the efficacy of statins would be imprudent, especially after repeatedly stressing that correlation doesn’t equal causation. It only further evinces their underlying motivations.
Next, Ravnskov et al. declare, “Also, the American National Health and Nutrition Examination Survey [103] found that during the period 1999–2006, the number of AMI and strokes increased from 3.4% to 3.7% and from 2.0% to 2.9%, respectively. During the same period, mean LDL-c level decreased from 126.1 to 114.8 mg/dl, and the self-reported use of lipid-lowering drugs increased from 8% to 13.4%.” Ravnskov et al. appear to insinuate that since LDL-c decreased and self-reported statin use increased, that the minor increase in stroke and AMI indicate statins are not effective at reducing CVD risk by lowering LDL-c. Yet again, they manage to completely dismiss or ignore many limitations that characterize this particular analysis78. Most importantly, the acute mean LDL-c values would not reflect chronic LDL-c of individuals in this sample. It is the cumulative LDL-c exposure that would be most relevant to the event rates in the seven years covered here. Even then, Kuklina et al. admitted they might have underestimated mean LDL-c levels by excluding long-term care facilities, which expands the disconnect between LDL-c and AMI/stroke event rates by itself. Also, data important to statin use was pretty scarce, leading them to directly state, “In addition, even though temporal changes in potential contributing factors, such as improvement in treatment of LDL-c levels, can be documented, the missing or limited information on the duration of treatment, dose titration, and changes in the type of medication used threatens the validity of such analyses.” Nonetheless, even if this information pertinent to dosing, treatment duration, and medication type was available and all of the previous issues did not exist, the decrease in mean LDL-c observed in the time frame studied was paltry at best. Such a minor difference likely would not have resulted in observable changes in CVD mortality unless all other risk factors were held constant for 10 to 20 years. However, this was certainly not what occurred during this time period, as here were obvious differences in other known CVD risk factors that could have explained the entirety of the slight increase in AMI and stroke rates (and subsequently the “lack of effect” of statin treatment).
At the very least, the higher diabetes prevalence and the slight increase in smoking rates would have been influential factors. Not to mention there was an abundance of other risk factors that weren’t even considered. Conveniently, Ravnskov et al. neglected to mention these factors and acted as if LDL-c was the only possible variable that could influence CVD risk. Ravnskov et al. also failed to acknowledge that a small ~10 mg/dL increase in LDL-c (due to minimal change in statin usage for those eligible for drug therapy) isn’t even expected to cause a major change in risk over seven years. A change, sure, but a larger difference in LDL-c or a more extended study duration would be needed to tease out the effects. I assume Ravnskov et al. purposefully ignore this point to imply that statins are not effective. Still, hopefully it goes without saying that the underlying assumptions and implications made here are asinine.
Carrying on with the same tenuous argument, Ravnskov et al. close the section off by saying, “Furthermore, statin utilization in 12 European countries between 2000 and 2012 was not associated with reduced CHD mortality or its rate of change over the years [104].” Commendably, this study actually chose a follow-up period a bit more capable of picking up potential effects on CVD mortality, but unfortunately, its strengths end there79. In the discussion section within the study, the authors (rightfully) addressed the main problems that Ravnskov et al. continually left out when attempting to use these publications to argue in their favor. The researchers noted numerous other risk factors for CVD that may have changed over the time period investigated, none of which were considered in their attempt to find a correlation. Thus, results from ecological studies are likely not applicable to individual patients as they lacked data on whether treatment was primary/secondary prevention, and the adherence rates to statins, and the doses used, and they even have little idea of whether the LDL-c levels were even achieved. These issues massively hinder the ability to reliably establish any correlation between statin use and CVD mortality that may exist. However, even though their analysis was plagued by a number of factors that would distort any correlation, their plot of change in annual statin use versus annual reductions in CHD mortality produced a distribution that wasn’t all that extraneous. The plot in reference showed a rough trend towards higher CHD mortality reductions with greater statin use changes. But don’t take this point too seriously because, in the end, using this to argue for or against the use of statins is a violation of the evidence hierarchy, and it would be foolish to do so.
Conclusion
Finally wrapping things up, in their concluding paragraph Ravnskov et al. proclaim, “The idea that high cholesterol levels in the blood are the main cause of CVD is impossible because people with low levels become just as atherosclerotic as people with high levels and their risk of suffering from CVD is the same or higher. The cholesterol hypothesis has been kept alive for decades by reviewers who have used misleading statistics, excluded the results from unsuccessful trials and ignored numerous contradictory observations.”
Impressively, they couldn’t even make it two sentences without making an illogical statement. As repeatedly demonstrated throughout the preceding discussion, the existence of groups that are more atherosclerotic despite having similar or lower LDL-c to comparator groups does not make it “impossible” that elevated serum LDL-c is the main cause of CVD. It simply illustrates that other known risk factors have additive effects on the risk of CVD, many of which are already well-characterized. Expectedly, those diagnosed with CVD in the analyses that Ravnskov et al. used to declare a lack of a relationship between LDL-c and CVD morbidity/mortality risk had much greater exposure to said factors alongside elevated serum LDL-c. This again showcases the selective ignorance of Ravnskov et al. to fit their narrative.
Moreover, their claims that the cholesterol hypothesis has been kept alive by reviewers using “misleading statistics” or who “excluded results from unsuccessful trials” and “ignored numerous contradictory observations” is unfounded. The statistics they called misleading were carried out exactly how they should be in the contexts they were used. In reality, Ravnskov et al. were the only ones using deceptive methods and failing to grasp fundamental principles of science. None of the trials they claimed was unsuccessful were excluded with malice, but rather excluded for perfectly valid reasons such as a small sample size, a short follow up regarding the outcome/s in question, the existence of competing risks, and low event rates that impaired statistical power to detect differences. Contradictory observations were also not ignored. The truth of the matter is that any “contradictory” observations were confined to low-quality analyses that either had blindingly apparent causes for the findings presented or were littered with methodological errors that stripped them of internal validity. Even if it were the case that contradictory observations were present, their mere existence does not invalidate the “cholesterol” (or really, lipid) hypothesis, and so to claim this is a logical error. The same concept applies to those who smoke and drink regularly yet don’t experience lung cancer, CVD, or other chronic conditions linked to the two; this does not disprove these activities cause these conditions. Like any competent group of scientists, the researchers who worked to establish the causal relationship between elevated LDL-c and CVD exercised caution in weighing results from lower tiers of the evidence hierarchy and only based their conclusions on consistent, comprehensive findings from robust, high-quality evidence. Factoring in all that has been discussed here, this review by Ravnskov et al. is merely a compilation of low-quality cherry-picked studies, misguided criticisms, logically invalid arguments, and scientific negligence. To advise against lipid-lowering therapy based on weak, inconsistent, and inconsequential criticisms offered by Ravnskov et al. is unbecoming and frankly dangerous. Doing so can take years off people’s lives.
Closing Notes
Although the following should be pretty evident, I want to emphasise one point to hopefully avoid the complaints, concerns, and commentary that often follow the release of these types of articles. While it is acknowledged that LDL-c, or more specifically apoB, is a causal factor for CVD, this does not mean that other risk factors do not exist and are not worth addressing. I tried to repeat this crucial point throughout the discussion. Attention should be given to any and all potential avenues of lifestyle modifications and/or medical treatment that can yield reductions in CVD risk. Likewise, my defense of the efficacy and safety of statins and other lipid-lowering drugs in primary and secondary CVD prevention is not an endorsement for unfettered recommendation and use of these therapies. They serve a specific purpose in select populations, but there are many nuances involving contraindications, effectiveness, side effects, and more that are best left for consideration by healthcare professionals. It is their job, not mine, to make an informed decision based on all the information at hand. All this breakdown intended to highlight is the misconstrual of evidence in Ravnskov et al.’s review, which continues to mislead readers and perpetuate health misinformation.
If you have enjoyed this article and are kind enough to support My Nutrition Science and its writers, please consider donating using the PayPal link here and signing up to our email list below.
SUBSCRIBE TO EMAIL LIST
References:
Ravnskov, Uffe, Michel de Lorgeril, David M. Diamond, Rokuro Hama, Tomohito Hamazaki, Björn Hammarskjöld, Niamh Hynes, et al. “LDL-c Does Not Cause Cardiovascular Disease: A Comprehensive Review of the Current Literature.” Expert Review of Clinical Pharmacology 11, no. 10 (October 3, 2018): 959–70. https://doi.org/10.1080/17512433.2018.1519391.
Ference, Brian A., Henry N. Ginsberg, Ian Graham, Kausik K. Ray, Chris J. Packard, Eric Bruckert, Robert A. Hegele, et al. “Low-Density Lipoproteins Cause Atherosclerotic Cardiovascular Disease. 1. Evidence from Genetic, Epidemiologic, and Clinical Studies. A Consensus Statement from the European Atherosclerosis Society Consensus Panel.” European Heart Journal 38, no. 32 (August 21, 2017): 2459–72. https://doi.org/10.1093/eurheartj/ehx144.
Lande, K. E., and W. M. Sperry. “Human atherosclerosis in relation to the cholesterol content of the blood serum.” Arch. Pathol. 22 (1936): 301–12.
Mathur, K. S., N. L. Patney, V. Kumar, and R. D. Sharma. “Serum Cholesterol and Atherosclerosis in Man.” Circulation 23, no. 6 (June 1, 1961): 847–52. https://doi.org/10.1161/01.CIR.23.6.847.
Paterson, J. C., Lucy Dyer, and E. C. Armstrong. “Serum Cholesterol Levels in Human Atherosclerosis.” Canadian Medical Association Journal 82, no. 1 (January 2, 1960): 6–11.
Marek, Zdzislaw, Kazimierz Jaegermann, and Tadeusz Ciba. “Atherosclerosis and Levels of Serum Cholesterol in Postmortem Investigations.” American Heart Journal 63, no. 6 (June 1, 1962): 768–74. https://doi.org/10.1016/0002-8703(62)90063-7.
Hecht, Harvey S, and H. Robert Superko. “Electron Beam Tomography and National Cholesterol Education Program Guidelines in Asymptomatic Women.” Journal of the American College of Cardiology 37, no. 6 (May 1, 2001): 1506–11. https://doi.org/10.1016/S0735-1097(01)01211-6.
Fernández-Friera, Leticia, Valentín Fuster, Beatriz López-Melgar, Belén Oliva, José M. García-Ruiz, José Mendiguren, Héctor Bueno, et al. “Normal LDL-cholesterol Levels Are Associated With Subclinical Atherosclerosis in the Absence of Risk Factors.” Journal of the American College of Cardiology 70, no. 24 (December 19, 2017): 2979–91. https://doi.org/10.1016/j.jacc.2017.10.024.
Sachdeva, Amit, Christopher P. Cannon, Prakash C. Deedwania, Kenneth A. Labresh, Sidney C. Smith, David Dai, Adrian Hernandez, and Gregg C. Fonarow. “Lipid Levels in Patients Hospitalized with Coronary Artery Disease: An Analysis of 136,905 Hospitalizations in Get With The Guidelines.” American Heart Journal 157, no. 1 (January 2009): 111-117.e2. https://doi.org/10.1016/j.ahj.2008.08.010.
Al-Mallah, Mouaz H., Hazem Hatahet, João L. Cavalcante, and Sanjaya Khanal. “Low Admission LDL-cholesterol Is Associated with Increased 3-Year All-Cause Mortality in Patients with Non ST Segment Elevation Myocardial Infarction.” Cardiology Journal 16, no. 3 (2009): 227–33.
Rott, David, Robert Klempfner, Ilan Goldenberg, and David Leibowitz. “Cholesterol Levels Decrease Soon after Acute Myocardial Infarction.” The Israel Medical Association Journal: IMAJ 17, no. 6 (June 2015): 370–73.
Kumar, Naresh, Suresh Kumar, Anil Kumar, Tariq Shakoor, and Amber Rizwan. “Lipid Profile of Patients with Acute Myocardial Infarction (AMI).” Cureus 11, no. 3 (n.d.): e4265. https://doi.org/10.7759/cureus.4265.
Ravnskov, U., McCully, K. S., & Rosch, P. J. (2012). The statin-low cholesterol-cancer conundrum. QJM : monthly journal of the Association of Physicians, 105(4), 383–388. https://doi.org/10.1093/qjmed/hcr243
Alsheikh-Ali, Alawi A., Thomas A. Trikalinos, David M. Kent, and Richard H. Karas. “Statins, Low-Density Lipoprotein Cholesterol, and Risk of Cancer.” Journal of the American College of Cardiology 52, no. 14 (September 30, 2008): 1141–47. https://doi.org/10.1016/j.jacc.2008.06.037.
Cholesterol Treatment Trialists’ (CTT) Collaboration, Jonathan R. Emberson, Patricia M. Kearney, Lisa Blackwell, Connie Newman, Christina Reith, Neeraj Bhala, et al. “Lack of Effect of Lowering LDL-c Cholesterol on Cancer: Meta-Analysis of Individual Data from 175,000 People in 27 Randomised Trials of Statin Therapy.” PloS One 7, no. 1 (2012): e29849. https://doi.org/10.1371/journal.pone.0029849.
Jeong, Gwang Hun, Keum Hwa Lee, Jong Yeob Kim, Michael Eisenhut, Andreas Kronbichler, Hans J. van der Vliet, Jae Il Shin, and Gabriele Gamerith. “Statin and Cancer Mortality and Survival: An Umbrella Systematic Review and Meta-Analysis.” Journal of Clinical Medicine 9, no. 2 (January 23, 2020): 326. https://doi.org/10.3390/jcm9020326.
Hoek, Hester L. van den, Willem Jan W. Bos, Anthonius de Boer, and Ewoudt M. W. van de Garde. “Statins and Prevention of Infections: Systematic Review and Meta-Analysis of Data from Large Randomised Placebo Controlled Trials.” BMJ 343 (November 29, 2011): d7281. https://doi.org/10.1136/bmj.d7281.
Feng, QiPing, Wei-Qi Wei, Sandip Chaugai, Barbara G. Carranza Leon, Jonathan D. Mosley, Daniel A. Carranza Leon, Lan Jiang, et al. “Association Between Low-Density Lipoprotein Cholesterol Levels and Risk for Sepsis Among Patients Admitted to the Hospital With Infection.” JAMA Network Open 2, no. 1 (January 18, 2019): e187223. https://doi.org/10.1001/jamanetworkopen.2018.7223.
Trinder, Mark, Keith R. Walley, John H. Boyd, and Liam R. Brunham. “Causal Inference for Genetically Determined Levels of High-Density Lipoprotein Cholesterol and Risk of Infectious Disease.” Arteriosclerosis, Thrombosis, and Vascular Biology 40, no. 1 (January 1, 2020): 267–78. https://doi.org/10.1161/ATVBAHA.119.313381.
Benn, Marianne, Anne Tybjærg-Hansen, Stefan Stender, Ruth Frikke-Schmidt, and Børge G. Nordestgaard. “Low-Density Lipoprotein Cholesterol and the Risk of Cancer: A Mendelian Randomization Study.” JNCI: Journal of the National Cancer Institute 103, no. 6 (March 16, 2011): 508–19. https://doi.org/10.1093/jnci/djr008.
Ravnskov, Uffe, David M Diamond, Rokura Hama, Tomohito Hamazaki, Björn Hammarskjöld, Niamh Hynes, Malcolm Kendrick, et al. “Lack of an Association or an Inverse Association between Low-Density-Lipoprotein Cholesterol and Mortality in the Elderly: A Systematic Review.” BMJ Open 6, no. 6 (June 2, 2016): e010401. https://doi.org/10.1136/bmjopen-2015-010401.
Bathum, Lise, René Depont Christensen, Lars Engers Pedersen, Palle Lyngsie Pedersen, John Larsen, and Jørgen Nexøe. “Association of Lipoprotein Levels with Mortality in Subjects Aged 50 + without Previous Diabetes or Cardiovascular Disease: A Population-Based Register Study.” Scandinavian Journal of Primary Health Care 31, no. 3 (September 1, 2013): 172–80. https://doi.org/10.3109/02813432.2013.824157.
Postmus, Iris, Joris Deelen, Sanaz Sedaghat, Stella Trompet, Anton J. M. de Craen, Bastiaan T. Heijmans, Oscar H. Franco, et al. “LDL-c Cholesterol Still a Problem in Old Age? A Mendelian Randomization Study.” International Journal of Epidemiology 44, no. 2 (April 2015): 604–12. https://doi.org/10.1093/ije/dyv031.
Richardson, Tom G., Qin Wang, Eleanor Sanderson, Anubha Mahajan, Mark I. McCarthy, Timothy M. Frayling, Mika Ala-Korpela, Allan Sniderman, George Davey Smith, and Michael V. Holmes. “Effects of Apolipoprotein B on Lifespan and Risks of Major Diseases Including Type 2 Diabetes: A Mendelian Randomisation Analysis Using Outcomes in First-Degree Relatives.” The Lancet Healthy Longevity 2, no. 6 (June 1, 2021): e317–26. https://doi.org/10.1016/S2666-7568(21)00086-6.
Burgess, Stephen, Nicholas J. Timpson, Shah Ebrahim, and George Davey Smith. “Mendelian Randomization: Where Are We Now and Where Are We Going?” International Journal of Epidemiology 44, no. 2 (April 2015): 379–88. https://doi.org/10.1093/ije/dyv108.
Tan, Yuan-De, Peng Xiao, and Chittibabu Guda. “In-Depth Mendelian Randomization Analysis of Causal Factors for Coronary Artery Disease.” Scientific Reports 10, no. 1 (June 8, 2020): 9208. https://doi.org/10.1038/s41598-020-66027-4.
“Influence of Pravastatin and Plasma Lipids on Clinical Events in the West of Scotland Coronary Prevention Study (WOSCOPS).” Circulation 97, no. 15 (April 21, 1998): 1440–45. https://doi.org/10.1161/01.CIR.97.15.1440.
Sacks, Frank M., Lemuel A. Moyé, Barry R. Davis, Thomas G. Cole, Jean L. Rouleau, David T. Nash, Marc A. Pfeffer, and Eugene Braunwald. “Relationship Between Plasma LDL-c Concentrations During Treatment With Pravastatin and Recurrent Coronary Events in the Cholesterol and Recurrent Events Trial.” Circulation 97, no. 15 (April 21, 1998): 1446–52. https://doi.org/10.1161/01.CIR.97.15.1446.
Schwartz, G. G., A. G. Olsson, M. D. Ezekowitz, P. Ganz, M. F. Oliver, D. Waters, A. Zeiher, et al. “Effects of Atorvastatin on Early Recurrent Ischemic Events in Acute Coronary Syndromes: The MIRACL Study: A Randomized Controlled Trial.” JAMA 285, no. 13 (April 4, 2001): 1711–18. https://doi.org/10.1001/jama.285.13.1711.
Silverman, Michael G., Brian A. Ference, Kyungah Im, Stephen D. Wiviott, Robert P. Giugliano, Scott M. Grundy, Eugene Braunwald, and Marc S. Sabatine. “Association Between Lowering LDL-c and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-Analysis.” JAMA 316, no. 12 (September 27, 2016): 1289. https://doi.org/10.1001/jama.2016.13985.
Abdullah, Shuaib M., Laura F. Defina, David Leonard, Carolyn E. Barlow, Nina B. Radford, Benjamin L. Willis, Anand Rohatgi, et al. “Long-Term Association of Low-Density Lipoprotein Cholesterol With Cardiovascular Mortality in Individuals at Low 10-Year Risk of Atherosclerotic Cardiovascular Disease.” Circulation 138, no. 21 (November 20, 2018): 2315–25. https://doi.org/10.1161/CIRCULATIONAHA.118.034273.
Li, Yingrui, Songbai Deng, Bin Liu, Yulin Yan, Jianlin Du, Yu Li, Xiaodong Jing, et al. “The Effects of Lipid-Lowering Therapy on Coronary Plaque Regression: A Systematic Review and Meta-Analysis.” Scientific Reports 11, no. 1 (April 12, 2021): 7999. https://doi.org/10.1038/s41598-021-87528-w.
Collins, Rory, Christina Reith, Jonathan Emberson, Jane Armitage, Colin Baigent, Lisa Blackwell, Roger Blumenthal, et al. “Interpretation of the Evidence for the Efficacy and Safety of Statin Therapy.” Lancet (London, England) 388, no. 10059 (November 19, 2016): 2532–61. https://doi.org/10.1016/S0140-6736(16)31357-5.
Collaboration, Cholesterol Treatment Trialists’. “Efficacy and Safety of LDL-c‐lowering Therapy among Men and Women: Meta‐analysis of Individual Data from 174,000 Participants in 27 Randomised Trials.” Accessed July 7, 2021. https://core.ac.uk/reader/41241600?utm_source=linkout.
Cannon, Christopher P., Eugene Braunwald, Carolyn H. McCabe, Daniel J. Rader, Jean L. Rouleau, Rene Belder, Steven V. Joyal, Karen A. Hill, Marc A. Pfeffer, and Allan M. Skene. “Intensive versus Moderate Lipid Lowering with Statins after Acute Coronary Syndromes.” Research-article. http://dx.doi.org/10.1056/NEJMoa040583. Massachusetts Medical Society, October 8, 2009. World. https://doi.org/10.1056/NEJMoa040583.
Koren, Michael J., and Donald B. Hunninghake. “Clinical Outcomes in Managed-Care Patients with Coronary Heart Disease Treated Aggressively in Lipid-Lowering Disease Management Clinics: The Alliance Study.” Journal of the American College of Cardiology 44, no. 9 (November 2, 2004): 1772–79. https://doi.org/10.1016/j.jacc.2004.07.053.
LaRosa, John C., Scott M. Grundy, David D. Waters, Charles Shear, Philip Barter, Jean-Charles Fruchart, Antonio M. Gotto, et al. “Intensive Lipid Lowering with Atorvastatin in Patients with Stable Coronary Disease.” The New England Journal of Medicine 352, no. 14 (April 7, 2005): 1425–35. https://doi.org/10.1056/NEJMoa050461.
Dujovne, Carlos A., A. N. Chremos, James L. Pool, Harold Schnaper, Reagan H. Bradford, Charles L. Shear, Jim Higgins, et al. “Expanded Clinical Evaluation of Lovastatin (EXCEL) Study Results: IV. Additional Perspectives on the Tolerability of Lovastatin.” The American Journal of Medicine 91, no. 1 (July 31, 1991): S25–30. https://doi.org/10.1016/0002-9343(91)90053-Z.
Kristensen, Malene Lopez, Palle Mark Christensen, and Jesper Hallas. “The Effect of Statins on Average Survival in Randomised Trials, an Analysis of End Point Postponement.” BMJ Open 5, no. 9 (September 24, 2015): e007118. https://doi.org/10.1136/bmjopen-2014-007118.
Finegold, Judith A., Matthew J. Shun-Shin, Graham D. Cole, Saman Zaman, Annette Maznyczka, Sameer Zaman, Rasha Al-Lamee, Siqin Ye, and Darrel P. Francis. “Distribution of Lifespan Gain from Primary Prevention Intervention.” Open Heart 3, no. 1 (March 1, 2016): e000343. https://doi.org/10.1136/openhrt-2015-000343.
Yebyo, Henock G, Hélène E Aschmann, Marco Kaufmann, and Milo A Puhan. “Comparative Effectiveness and Safety of Statins as a Class and of Specific Statins for Primary Prevention of Cardiovascular Disease: A Systematic Review, Meta-Analysis, and Network Meta-Analysis of Randomized Trials with 94,283 Participants.” American Heart Journal 210 (April 1, 2019): 18–28. https://doi.org/10.1016/j.ahj.2018.12.007.
Kjekshus, John, Eduard Apetrei, Vivencio Barrios, Michael Böhm, John G. F. Cleland, Jan H. Cornel, Peter Dunselman, et al. “Rosuvastatin in Older Patients with Systolic Heart Failure.” Research-article. http://dx.doi.org/10.1056/NEJMoa0706201. Massachusetts Medical Society, October 9, 2009. World. https://doi.org/10.1056/NEJMoa0706201.
Koren, Michael J., and Donald B. Hunninghake. “Clinical Outcomes in Managed-Care Patients with Coronary Heart Disease Treated Aggressively in Lipid-Lowering Disease Management Clinics: The Alliance Study.” Journal of the American College of Cardiology 44, no. 9 (November 2, 2004): 1772–79. https://doi.org/10.1016/j.jacc.2004.07.053.
Deedwania, Prakash, Peter H. Stone, C. Noel Bairey Merz, Juan Cosin-Aguilar, Nevres Koylan, Don Luo, Pamela Ouyang, et al. “Effects of Intensive Versus Moderate Lipid-Lowering Therapy on Myocardial Ischemia in Older Patients With Coronary Heart Disease.” Circulation 115, no. 6 (February 13, 2007): 700–707. https://doi.org/10.1161/CIRCULATIONAHA.106.654756.
Pedersen, Terje R., Ole Faergeman, John J. P. Kastelein, Anders G. Olsson, Matti J. Tikkanen, Ingar Holme, Mogens Lytken Larsen, et al. “High-Dose Atorvastatin vs Usual-Dose Simvastatin for Secondary Prevention after Myocardial Infarction: The IDEAL Study: A Randomized Controlled Trial.” JAMA 294, no. 19 (November 16, 2005): 2437–45. https://doi.org/10.1001/jama.294.19.2437.
Sathasivam, S., and B. Lecky. “Statin Induced Myopathy.” BMJ 337, no. nov06 3 (November 6, 2008): a2286–a2286. https://doi.org/10.1136/bmj.a2286.
Phillips, Paul S., Richard H. Haas, Sergei Bannykh, Stephanie Hathaway, Nancy L. Gray, Bruce J. Kimura, Georgirene D. Vladutiu, and John D.F. England. “Statin-Associated Myopathy with Normal Creatine Kinase Levels.” Annals of Internal Medicine 137, no. 7 (October 1, 2002): 581–85. https://doi.org/10.7326/0003-4819-137-7-200210010-00009.
Sinzinger, H, and J O’Grady. “Professional Athletes Suffering from Familial Hypercholesterolaemia Rarely Tolerate Statin Treatment Because of Muscular Problems.” British Journal of Clinical Pharmacology 57, no. 4 (April 2004): 525–28. https://doi.org/10.1111/j.1365-2125.2004.02044.x.
Wood, Frances A., James P. Howard, Judith A. Finegold, Alexandra N. Nowbar, David M. Thompson, Ahran D. Arnold, Christopher A. Rajkumar, et al. “N-of-1 Trial of a Statin, Placebo, or No Treatment to Assess Side Effects.” New England Journal of Medicine 383, no. 22 (November 26, 2020): 2182–84. https://doi.org/10.1056/NEJMc2031173.
Yatham, Megha Sai, Kavya Sai Yatham, Arun V. Ravindran, and Frank Sullivan. “Do Statins Have an Effect on Depressive Symptoms? A Systematic Review and Meta-Analysis.” Journal of Affective Disorders 257 (October 1, 2019): 55–63. https://doi.org/10.1016/j.jad.2019.07.002.
Chu, Che-Sheng, Ping-Tao Tseng, Brendon Stubbs, Tien-Yu Chen, Chia-Hung Tang, Dian-Jeng Li, Wei-Cheng Yang, et al. “Use of Statins and the Risk of Dementia and Mild Cognitive Impairment: A Systematic Review and Meta-Analysis.” Scientific Reports 8, no. 1 (April 11, 2018): 5804. https://doi.org/10.1038/s41598-018-24248-8.
Poly, Tahmina Nasrin, Md Mohaimenul Islam, Bruno Andreas Walther, Hsuan-Chia Yang, Chieh-Chen Wu, Ming-Chin Lin, and Yu-Chuan Li. “Association between Use of Statin and Risk of Dementia: A Meta-Analysis of Observational Studies.” Neuroepidemiology 54, no. 3 (2020): 214–26. https://doi.org/10.1159/000503105.
Ott, Brian R., Lori A. Daiello, Issa J. Dahabreh, Beth A. Springate, Kimberly Bixby, Manjari Murali, and Thomas A. Trikalinos. “Do Statins Impair Cognition? A Systematic Review and Meta-Analysis of Randomized Controlled Trials.” Journal of General Internal Medicine 30, no. 3 (March 2015): 348–58. https://doi.org/10.1007/s11606-014-3115-3.
Yu, Shandong, Yanpeng Chu, Gang Li, Lu Ren, Qing Zhang, and Lin Wu. “Statin Use and the Risk of Cataracts: A Systematic Review and Meta‐Analysis.” Journal of the American Heart Association 6, no. 3 (n.d.): e004180. https://doi.org/10.1161/JAHA.116.004180.
Yan, Junqiang, Liang Qiao, Jing Tian, Anran Liu, Jiannan Wu, Jiarui Huang, Mengmeng Shen, and Xiaoyi Lai. “Effect of Statins on Parkinson’s Disease: A Systematic Review and Meta-Analysis.” Medicine 98, no. 12 (March 2019): e14852. https://doi.org/10.1097/MD.0000000000014852.
Wang, Miao, Muqin Li, and Ying Xie. “The Association between Statins Exposure and Peripheral Neuropathy Risk: A Meta-Analysis.” Journal of Clinical Pharmacy and Therapeutics 46, no. 4 (August 2021): 1046–54. https://doi.org/10.1111/jcpt.13393.
Elgendy, Akram Y., Islam Y. Elgendy, Ahmed N. Mahmoud, Mohammad Al-Ani, Mohamed Moussa, Ahmad Mahmoud, Mohammad K. Mojadidi, and R. David Anderson. “Statin Use in Men and New Onset of Erectile Dysfunction: A Systematic Review and Meta-Analysis.” The American Journal of Medicine 131, no. 4 (April 1, 2018): 387–94. https://doi.org/10.1016/j.amjmed.2017.10.043.
Cai, Xiang, Ye Tian, Tao Wu, Chen-Xi Cao, Si-Yuan Bu, and Kun-Jie Wang. “The Role of Statins in Erectile Dysfunction: A Systematic Review and Meta-Analysis.” Asian Journal of Andrology 16, no. 3 (June 2014): 461–66. https://doi.org/10.4103/1008-682X.123678.
Kostis, John B., and Jeanne M. Dobrzynski. “The Effect of Statins on Erectile Dysfunction: A Meta‐Analysis of Randomized Trials.” The Journal of Sexual Medicine 11, no. 7 (July 1, 2014): 1626–35. https://doi.org/10.1111/jsm.12521.
Fan, Lailai, Yangyang Wang, Xiang Liu, and Xueqiang Guan. “Association between Statin Use and Herpes Zoster: Systematic Review and Meta-Analysis.” BMJ Open 9, no. 2 (February 1, 2019): e022897. https://doi.org/10.1136/bmjopen-2018-022897.
Thakker, Divyesh, Sunita Nair, Amit Pagada, Vinayak Jamdade, and Anuradha Malik. “Statin Use and the Risk of Developing Diabetes: A Network Meta-Analysis.” Pharmacoepidemiology and Drug Safety 25, no. 10 (October 2016): 1131–49. https://doi.org/10.1002/pds.4020.
Cohen, David E. “Balancing Cholesterol Synthesis and Absorption in the Gastrointestinal Tract.” Journal of Clinical Lipidology 2, no. 2 (April 2008): S1–3. https://doi.org/10.1016/j.jacl.2008.01.004.
Karagiannis, Angelos D, Anurag Mehta, Devinder S Dhindsa, Salim S Virani, Carl E Orringer, Roger S Blumenthal, Neil J Stone, and Laurence S Sperling. “How Low Is Safe? The Frontier of Very Low (<30 Mg/DL) LDL-c Cholesterol.” European Heart Journal 42, no. 22 (June 7, 2021): 2154–69. https://doi.org/10.1093/eurheartj/ehaa1080.
Leenaars, Cathalijn H. C., Carien Kouwenaar, Frans R. Stafleu, André Bleich, Merel Ritskes-Hoitinga, Rob B. M. De Vries, and Franck L. B. Meijboom. “Animal to Human Translation: A Systematic Scoping Review of Reported Concordance Rates.” Journal of Translational Medicine 17, no. 1 (July 15, 2019): 223. https://doi.org/10.1186/s12967-019-1976-2.
Sabatine, Marc S., Robert P. Giugliano, Anthony C. Keech, Narimon Honarpour, Stephen D. Wiviott, Sabina A. Murphy, Julia F. Kuder, et al. “Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease.” Research-article. http://dx.doi.org/10.1056/NEJMoa1615664. Massachusetts Medical Society, March 17, 2017. World. https://doi.org/10.1056/NEJMoa1615664.
Schmidt, Amand F., John-Paul L. Carter, Lucy S. Pearce, John T. Wilkins, John P. Overington, Aroon D. Hingorani, and J. P. Casas. “PCSK9 Monoclonal Antibodies for the Primary and Secondary Prevention of Cardiovascular Disease.” The Cochrane Database of Systematic Reviews 10 (October 20, 2020): CD011748. https://doi.org/10.1002/14651858.CD011748.pub3.
“Risk of Fatal Coronary Heart Disease in Familial Hypercholesterolaemia. Scientific Steering Committee on Behalf of the Simon Broome Register Group.” BMJ : British Medical Journal 303, no. 6807 (October 12, 1991): 893–96.
Ho, Anthony M.-H., Peter W. Dion, Janice H. H. Yeung, John B. Holcomb, Lester A. H. Critchley, Calvin S. H. Ng, Manoj K. Karmakar, Chi W. Cheung, Timothy H. Rainer, and David S. Warner. “Prevalence of Survivor Bias in Observational Studies on Fresh Frozen Plasma: Erythrocyte Ratios in Trauma Requiring Massive Transfusion.” Anesthesiology 116, no. 3 (March 1, 2012): 716–28. https://doi.org/10.1097/ALN.0b013e318245c47b.
Mundal, Liv, Mirza Sarancic, Leiv Ose, Per Ole Iversen, Jens‐Kristian Borgan, Marit B. Veierød, Trond P. Leren, and Kjetil Retterstøl. “Mortality Among Patients With Familial Hypercholesterolemia: A Registry‐Based Study in Norway, 1992–2010.” Journal of the American Heart Association 3, no. 6 (n.d.): e001236. https://doi.org/10.1161/JAHA.114.001236.
Seed, Mary, Fritz Hoppichler, David Reaveley, Susan McCarthy, Gilbert R. Thompson, Eric Boerwinkle, and Gerd Utermann. “Relation of Serum Lipoprotein(a) Concentration and Apolipoprotein(a) Phenotype to Coronary Heart Disease in Patients with Familial Hypercholesterolemia.” Research-article. http://dx.doi.org/10.1056/NEJM199005243222104. Massachusetts Medical Society, January 14, 2010. World. https://doi.org/10.1056/NEJM199005243222104.
Wiklund, O., B. Angelin, S. O. Olofsson, M. Eriksson, G. Fager, L. Berglund, and G. Bondjers. “Apolipoprotein(a) and Ischaemic Heart Disease in Familial Hypercholesterolaemia.” Lancet (London, England) 335, no. 8702 (June 9, 1990): 1360–63. https://doi.org/10.1016/0140-6736(90)91242-3.
Vuorio, A. F., H. Turtola, K.-M. Piilahti, P. Repo, T. Kanninen, and K. Kontula. “Familial Hypercholesterolemia in the Finnish North Karelia.” Arteriosclerosis, Thrombosis, and Vascular Biology 17, no. 11 (November 1, 1997): 3127–38. https://doi.org/10.1161/01.ATV.17.11.3127.
Wittekoek, M. E., E. de Groot, M. H. Prins, M. D. Trip, H. R. Büller, and J. J. Kastelein. “Differences in Intima-Media Thickness in the Carotid and Femoral Arteries in Familial Hypercholesterolemic Heterozygotes with and without Clinical Manifestations of Cardiovascular Disease.” Atherosclerosis 146, no. 2 (October 1999): 271–79. https://doi.org/10.1016/s0021-9150(99)00147-1.
Smilde, T. J., M. D. Trip, H. Wollersheim, S. van Wissen, J. J. Kastelein, and A. F. Stalenhoef. “Rationale, Design and Baseline Characteristics of a Clinical Trial Comparing the Effects of Robust vs Conventional Cholesterol Lowering and Intima Media Thickness in Patients with Familial Hypercholesterolaemia : The Atorvastatin versus Simvastatin on Atherosclerosis Progression (ASAP) Study.” Clinical Drug Investigation 20, no. 2 (2000): 67–79. https://doi.org/10.2165/00044011-200020020-00001.
Cenarro, A., M. Artieda, S. Castillo, P. Mozas, G. Reyes, D. Tejedor, R. Alonso, et al. “A Common Variant in the ABCA1 Gene Is Associated with a Lower Risk for Premature Coronary Heart Disease in Familial Hypercholesterolaemia.” Journal of Medical Genetics 40, no. 3 (March 2003): 163–68. https://doi.org/10.1136/jmg.40.3.163.
Nilsson, Staffan, Sigvard Mölstad, Catarina Karlberg, Jan-Erik Karlsson, and Lars-Göran Persson. “No Connection between the Level of Exposition to Statins in the Population and the Incidence/Mortality of Acute Myocardial Infarction: An Ecological Study Based on Sweden’s Municipalities.” Journal of Negative Results in Biomedicine 10 (May 24, 2011): 6. https://doi.org/10.1186/1477-5751-10-6.
Kuklina, Elena V. “Trends in High Levels of Low-Density Lipoprotein Cholesterol in the United States, 1999-2006.” JAMA 302, no. 19 (November 18, 2009): 2104. https://doi.org/10.1001/jama.2009.1672.
Vancheri, Federico, Lars Backlund, Lars-Erik Strender, Brian Godman, and Björn Wettermark. “Time Trends in Statin Utilisation and Coronary Mortality in Western European Countries.” BMJ Open 6, no. 3 (March 30, 2016): e010500. https://doi.org/10.1136/bmjopen-2015-010500.
Related Deep Dives
Bodyweight Control
Shaun Ward
Eating Behaviour and Obesity are Choices? When Science Meets Philosophy
Disease Prevention
Shaun Ward
Merging Reductionism and Holism in Nutrition
Disease Prevention
Shaun Ward
Fibre and Constipation: Correcting Paul Mason Misinformation





























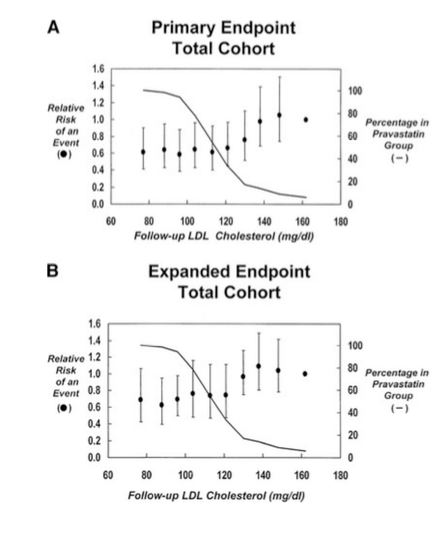 They also determined that the absolute LDL-c reduction (as a numerical value or a percentage) was not a significant predictor of CVD mortality/morbidity risk reduction.
They also determined that the absolute LDL-c reduction (as a numerical value or a percentage) was not a significant predictor of CVD mortality/morbidity risk reduction. 























 Section 6.4
Section 6.4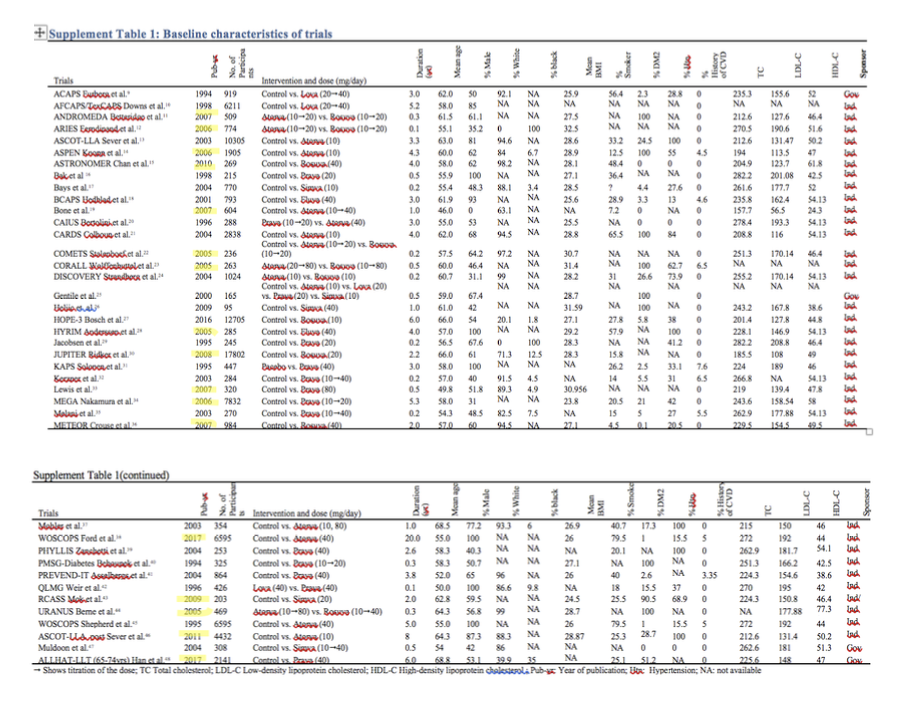

 Section 6.4
Section 6.4




 So, in addition to the fact it appears perfectly reasonable to expect nocebo effects may comprise a non-negligible proportion of subjective statin-related side effects such as myopathy, nothing Ravnskov et al. bring to the table changes the validity of Collins et al. statements about the frequency of this type of side effect. The entire thing is a complete non-sequitur. The information that Ravnskov et al. share here just indicates that there may be certain subsets of the population who experience rarer, novel side-effects and therefore require different treatments for elevated LDL-c. None of the authors of these publications deny this, and it doesn’t disprove anything previously stated. It only seems as though this was another concerted effort by Ravnskov et al. to inappropriately dissuade individuals from starting a statin if one is advised.
So, in addition to the fact it appears perfectly reasonable to expect nocebo effects may comprise a non-negligible proportion of subjective statin-related side effects such as myopathy, nothing Ravnskov et al. bring to the table changes the validity of Collins et al. statements about the frequency of this type of side effect. The entire thing is a complete non-sequitur. The information that Ravnskov et al. share here just indicates that there may be certain subsets of the population who experience rarer, novel side-effects and therefore require different treatments for elevated LDL-c. None of the authors of these publications deny this, and it doesn’t disprove anything previously stated. It only seems as though this was another concerted effort by Ravnskov et al. to inappropriately dissuade individuals from starting a statin if one is advised.